Baeckvall J.-E. (ed.) Modern Oxidation Methods
Подождите немного. Документ загружается.

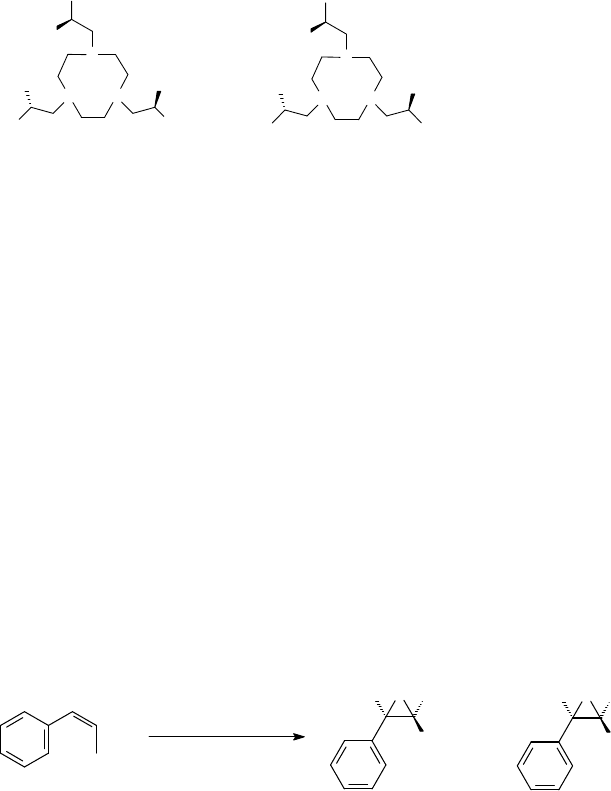
Enantioselective Alkene Epoxidation Although ascorbic acid is chiral and plays a key
role in boosting the activity of 6, enantioselective epoxidation was not observed [94a].
By contrast, Beller, Bolm, and coworkers reported that enantiomerically enriched
epoxides were obtained with manganese complexes based on chiral analogs of the
tmtacn ligand (Figure 11.8) [94f,103,104]. The first asymmetric epoxidation reactions
were catalyzed by in situ prepared Mn catalysts from N-substituted chiral tacn ligands
[103a]. The chirality was introduced via alkylation of the secondary amine moieties
to generate the C
3
-symmetric ligands depicted in Figure 11.8.
Styrene was converted to the corresponding epoxide with an ee of 43%, albeit in
only 15% yield after 5 h using H
2
O
2
and the Mn complex based on ligand 25. Using
longer reaction times, higher temperatures and higher catalyst loadings, the yield
was increased but at the price of a decrease in enantioselectivity [103a]. With the
sterically more demanding ligand 26, enantioselectivities in the range 13–38% were
observed for styrene and chromene. Higher enantioselectivity was achieved with
cis-b-methylstyrene as substrate. The trans-epoxide was found as the major product in
55% ee whereas the cis-epoxide was formed as the minor product with an ee of 13%
(Scheme 11.12).
The enantiopure C
3
-symmetric tris-pyrrolidine-1,4,7-triazacyclononane ligand 27
has been reported by Bolm and coworkers (Figure 11.9) [94f]. The tacn derivative
was obtained by reduction of an
L-proline-derived cyclotripeptide, and the corre-
sponding dinuclear manganese complex was applied in the catalytic enantioselective
epoxidation of vinylarenes with H
2
O
2
as oxidant. For the epoxidation of styrene,
3-nitrostyrene, and 4-chlorostyrene excellent conversions (up to 88%) and ees of up to
30% were obtained [94f]. In addition to the chiral C
3
- and C
1
-symmetric ligands,
NN
N
iPr
iPr iP
r
HO OH
HO
NN
N
Me
eMeM
HO OH
HO
26
25
Figure 11.8 C
3
-symmetric chiral ligands [103a].
H
CH
3
HO
CH
3
HHO
3 mol% Mn(OAc)
2
.4H
2
O
4.5 mol% ligand
2 equiv. H
2
O
2
(aq. 30%)
MeOH, 0
o
C
+
55% e.e.
26
Scheme 11.12 Asymmetric epoxidation with Mn complex of ligand 26 [103a].
11.4 Epoxidation and cis-Dihydroxylation of Alkenes
j
393
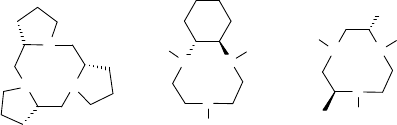
C
2
-symmetric tacn analogs have been tested (Figure 11.9) [103]. Thus far, modest
enantioselectivity has been obtained, but the potential of these tmtacn analogs by
further fine-tuning of the chiral ligand structure is evident.
Recently S
€
uss-Fink, Schulpin and coworkers have reported two tmtacn detivatives
where one of the methyl groups has been replaced by a 2-methyl-butyl or 2-hydro-
xybutyl group [105]. The catalytic activity was examined in the oxidation of indene
and phenylethanol (racemic) in the presence of ascorbic acid [94] or oxalate [97].
Although moderate activity was observed (up to 300 t.o.n.), unfortunately only very
low levels of enantioselectivity were achieved (<17%).
Kilic et al. have reported an alternative approach to controlling the stereochemical
outcome of the epoxidation catalyzed by Mn-tmtacn complexes using substrate
control via the hydroxyl group of allylic alcohols and by varying the steric nature of the
substituents on the alkene [106].
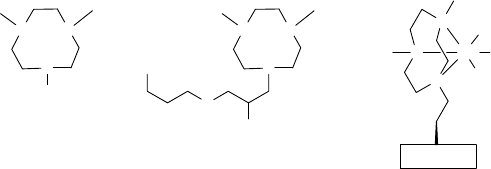
Manganese-tacn Derivatives on Solid Supports Several successful attempts to im-
prove catalyst selectivity have been made by encapsulation of the Mn-tmtacn complex
in zeolites [107]. Immobilization of the triazacyclononane ligand on an inorganic
support provided a new class of heterogeneous manganese catalysts with increased
epoxidation selectivity [108]. A novel approach to immobilizing Mn-tmacn-based
catalysts has been taken by Veghini et al. who employed tungstosilicic acid (as a large
anion) to render the catalyst insoluble but still allowing it to be dispersed in
solution [109]. However, as is often the case with immobilized homogenous catalysts,
the conversions were lower than those obtained with the analogous homogenous
catalysts [108].
A notable exception to this is found in the seminal work of De Vos and Jacobs on
immobilization of the tmtacn catalyst onto silica. The heterogenization procedure of
the tacn ligand started with the conversion of dimethyl tacn (dmtacn, 30, Figure 11.10)
to the silylated compound 31 with 3-(glycidyloxy)propyltrimethoxysilane followed
by immobilization on an SiO
2
surface and subsequently metalation of the hetero-
genized ligand with MnSO
4
.H
2
O [99]. Not only was this system highly effective, but
serendipitously it led to the discovery of a previously unexplored field of oxidation
catalysis for the tmtacn family of catalysts, that of cis-dihydroxylation of alkenes.
This will be discussed in more depth in the following section. A similar approach to
NN
N
Me
Me
Me
NN
N
NN
N
Me
Me
Me
R
2
R
1
R
1
= R
2
= Me
R
1
= R
2
=
i
Pr
27
28 29
Figure 11.9 Chiral tmtacn based ligands.
394
j
11 Manganese-Catalyzed Oxidation with Hydrogen Peroxide
immobilizing manganese polypyridyl catalysts has been taken by Stack and
coworkers recently, which may be of relevance to the tmtacn-based systems [110].
By using a copper templating approach, the immobilization of the ligands in
sufficient proximity to allow for two ligands to bind each manganese ion was found
to be essential to achieve full catalytic activity. Although the system employed
peracetic acid as the oxidant, it nevertheless is of direct relevance to the future
design of immobilized manganese-based oxidation catalysts. For example, where a
binuclear catalyst is the active species under homogenous conditions, low surface
coverage of the ligand will limit the ability to form such catalysts in a heterogenized
form. The templating approach allows for two ligands to be immobilized in proximity
to each other and thereby overcomes this problem.
cis-Dihydroxylation of Alkenes cis-Dihydroxylation is an important synthetic trans-
formation, and several reagents can be used for the addition of two hydroxyl groups to
an alkene. Both OsO
4
and alkaline KMnO
4
are suitable for cis-dihydroxylation [1, 14],
but the catalytic versions of the OsO
4
method using O
2
[111], H
2
O
2
[112], or other
oxidants [113] is the method of choice for this transformation [114]. The introduction
of the highly enantioselective OsO
4
-catalyzed dihydroxylation by Sharpless and its
extensive use in synthetic chemistry in recent years have provided ample demon-
stration of the key role of this oxidation reaction [115]. The toxicity of OsO
4
and the
stoichiometric nature of the KMnO
4
cis-dihydroxylation, which usually provides only
modest yields of diols, are strong incentives to develop catalytic cis-dihydroxylation
reactions with H
2
O
2
such as those based on Fe [116] and Mn (see below).
When using the heterogenized Mn-tmtacn system for the oxidation of alkenes (see
above), De Vos, Jacobs and coworker noted that substantial amounts of cis-diol were
formed in addition to the expected epoxide [99]. In oxidation reactions with 32, with
H
2
O
2
as oxidant and CH
3
CN as solvent, alkenes were converted to the corresponding
cis-diols albeit with the catalyst activity with respect to cis-diol formation being modest
(10–60 mol cis-diol/mol Mn) and epoxides being the major products. For internal
alkenes, for example, 2-hexene, retention of configuration was found for both
epoxide and cis-diol. Control experiments with dmtacn 30 showed severe peroxide
decomposition, and no oxidation products were obtained. A sufficiently long-lived
MnN
N
N
X
OH
2
OH
2
SiO
2
NN
N
OH
O
Si(OCH
3
)
3
NN
N
H
30 31
32
Figure 11.10 Structures of dmtacn (30), heterogenizable ligand 31, and the proposed active
complex 32 (X ¼ activated O to be transferred) [99].
11.4 Epoxidation and cis-Dihydroxylation of Alkenes
j
395
mononuclear complex (32) was postulated as the active species for both epoxidation
and cis-dihydroxylation. This complex contains cis coordination sites for labile
ligands (e.g., H
2
O and X), and both oxygen atoms from H
2
O and X (the activated
oxygen) are proposed to be transferred to the alkene to produce the cis-diol [99].
Although the proposed structure was speculative, it did correspond with the results
obtained experimentally.
Prompted by the use of oxalate and other additives and by the observation of cis-diol
formation in the hetereogenized system of De Vos and coworkers, a wider search
for effective additives both to suppress the catalase type activity of the Mn-tmtacn
complexes and to promote cis-dihydroxylation was initiated. An early success was the
strongly enhanced cis-dihydroxylation activity observed using [Mn
2
O
3
(tmtacn)
2
]
(PF
6
)
2
(6) (Figure 11.2) in the combination with aldehydes such as glyoxylic acid
methylester methyl hemiacetal (gmha (33), Scheme 11.13) [94k].
This mixed Mn-tmtacn/activated carbonyl system provided for a highly active and
H
2
O
2
efficient catalyst for the epoxidation of alkenes as well as the first homogeneous
Mn-based catalytic cis-dihydroxylation system using H
2
O
2
. Catalytic oxidations were
performed by slow addition of aqueous 50% H
2
O
2
(1.3 equiv. with respect to the
substrate) to a mixture of alkene, the Mn-tmtacn catalyst (0.1 mol%), and gmha
(25 mol%) in CH
3
CN at 0
C. Under these reaction conditions high conversions are
reached, whereas only 30% excess of oxidant with respect to substrate was required
(Table 11.8). The H
2
O
2
efficiency represented a dramatic improvement compared to
previous Mn-based systems [132]. In most cases the conversions were also signif-
icantly higher than those obtained with oxalate as co-catalyst using 1.3 equiv. of H
2
O
2
.
Substantial amounts of cis-diols were formed with the cis-diol/epoxide ratio depend-
ing heavily on the alkene structure. The highest selectivity for cis-dihydroxylation was
obtained with cyclooctene (Scheme 11.13), which afforded the cis-diol as the main
product (42%, 420 t.o.n.). Minor amounts of 2-hydroxycyclooctanone were observed
due to overoxidation of the diol. The ring size of cycloalkenes had a profound
influence on the cis-diol/epoxide product ratio. For cyclic alkenes, almost no trans-diol
could be detected (ratio cis-diol/trans-diol >99.5/0.5). cis-Diol formation is also
observed for aliphatic acyclic alkenes. Yields of diol were significantly lower for
trans-2-hexene than for cis-2-hexene, but the cis-diol/epoxide ratio was similar for both
substrates. The aryl-substituted alkenes yielded nearly exclusively epoxide under
these conditions. Importantly, diols were not formed when the substrate was replaced
by the corresponding epoxide in the reaction, excluding epoxide hydrolysis as a
source of diol.
Since cis-diol formation through Mn-catalyzed epoxide hydrolysis can be excluded
it was proposed that the cis-diols were formed by reaction of the alkene with a Mn oxo-
hydroxo species. As in the case of oxalate, activated carbonyl compounds such as
gmha
1)
could in principle inhibit the catalase-active dinuclear Mn complex 6
(Figure 11.2) [34] through formation of mononuclear Mn species via complexation
1) Gmha is an equilibrium mixture, which also contains some hydrated methyl glyoxylate. NMR
experiments indicated that the formation peroxyhydrate from gmha and aqueous H
2
O
2
is established
slowly.
396
j
11 Manganese-Catalyzed Oxidation with Hydrogen Peroxide
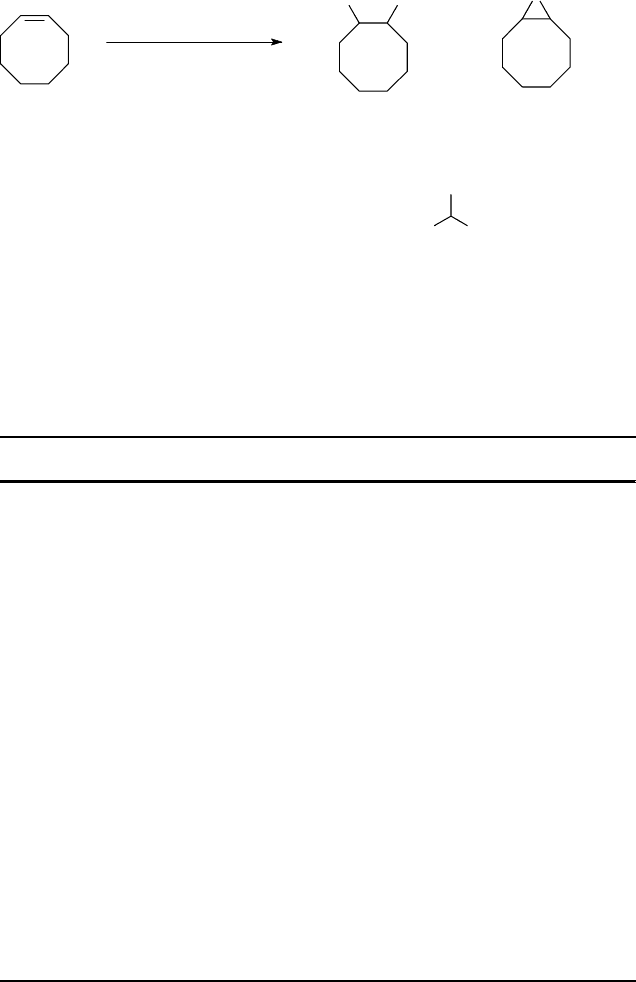
Table 11.8 cis-Dihydroxylation and epoxidation of selected alkenes by H
2
O
2
with Mn-tmtacn/gmha
catalysts [132].
Substrate Conversion % Product Turnover number (t.o.n.)
a)
Cyclohexene 88 Epoxide 590
cis-Diol 90
2-Cyclohexenone 80
Cyclooctene 90 Epoxide 360
cis-Diol 420
2-HO-Cyclooctanone 220
Norbornylene 95 exo Epoxide 540
exo-cis-Diol 180
trans-2-Hexene 77 trans-Epoxide 210
cis-Epoxide 50
RR/SS-Diol 150
RS/SR-Diol 0
cis-2-Hexene 93 cis-Epoxide 450
trans-Epoxide 40
SR/RS-Diol 280
RR/SS-Diol 10
cis-Stilbene 82 cis-Epoxide 260
trans-Epoxide 200
meso-Hydrobenzoin 40
Hydrobenzoin 40
Styrene 97 Epoxide 860
Ph(CH)(OH)CH
2
OH 60
PhC(O)CH
2
OH 10
a) 50% aqueous H
2
O
2
(1.3 equiv. with respect to substrate) was added over 6 h to a mixture of alkene
(40 mmol), Mn
2
O
3
(tmtacn)
2
(PF
6
)
2
(0.1 mol%), and GMHA (25 mol%) in MeCN (40 ml) with
internal standard (1.2-dichlorobenzene, 20 mmol) at 0
C. Analysis by GC 1 h after addition of
oxidant was completed.
0.1 mol%
25 mol% gmha
1.3 equiv. H
2
O
2
(aq. 50 %)
MeCN
O
OHHO
+
Cis-diol
42% 36%
gmha =
OCH
3
CO
2
CH
3
HO
6
33
(33)
Scheme 11.13 Dihydroxylation of alkenes in the presence of glyoxylic acid methyl ester methyl
hemiacetal (33) [94k].
11.4 Epoxidation and cis-Dihydroxylation of Alkenes
j
397
to the Mn center. cis-Diol formation from an Mn oxo-hydroxo species with a
coordinated hydrated carbonyl ligand could be induced through a hydrogen-bonded
6- membered ring transition state (concerted pathway, Scheme 11.14). Reoxidation of
the Mn center by H
2
O
2
, release of the diol from Mn, and hydration of the carbonyl
compound closes the catalytic cycle. It should be noted that this mechanism was
tentative and based on general principles of manganese coordination chemistry
rather than empirical data, which, once it became available (see below), as is so often
the case, demonstrated that a thorough understanding of a system is essential to
unraveling mechanistic aspects. Nevertheless, the use of activated carbonyl com-
pounds in combination with Mn-tmtacn not only provides for a highly active (up to
860 t.o.n.) and H
2
O
2
-efficient epoxidation system (see above), but also was the most
active Os-free homogeneous catalyst for cis-dihydroxylation (up to 420 t.o.n.),
reported at that stage. Due to competing cis-dihydroxylation and epoxidation path-
ways it was not suitable for routine application in synthesis, however. Nevertheless it
proved an important lead to develop highly selective Mn-catalyzed cis-dihydroxylation
systems employing H
2
O
2
.
Subsequent to these re ports on the activity of aldehydes in enhancing the
reactivity of Mn-tmtacn-based catalysts, Feringa and coworkers demonstrated that
carboxylic acids at co-catalytic levels wer eactuallyresponsiblefortheenhancement,
and that the aldehydes served solely as a source of these acids [117]. Indeed, in the
presence of carboxylic acids, complex 6 [M n
IV
2
(m-O)
3
(tmtacn)
2
]
2 þ
proved to be
highly efficient in catalyzing the oxidation of alkenes to the corresponding ci s -diol
and epoxide products using H
2
O
2
. The selectivity of the catal ytic syst em both in
terms of cis-dihydroxylation and epoxidation of alkenes could be tuned readily by
judicious choice of the carboxylic acid employed. High turnover numbers (t.o.n.
cis-alkene
cis
concerted pathway
O
O
H
O
Mn
OL
X
H
*
O
O
H
O
Mn
OL
X
H
*
O
O
H
O
Mn
OL
X
H
Mn
L
O
O
+
H
2
O
O
HX
+
33
34
35
Scheme 11.14 Early proposal for the mechanism of cis-dihydroxylation by 6 (L ¼ tmtacn,
X ¼ CO
2
Me). Note that gmha is an equilibrium mixture which also contains some hydrated methyl
glyoxylate.
398
j
11 Manganese-Catalyzed Oxidation with Hydrogen Peroxide
>2000) were achie ved, especially for cis-dihydroxylation, that is, with 2,6-dichloro-
benzoic acid, the highest t.o.n. reported thus far for cis-dihydroxylation of alkenes
catalyzed by an osmium free system. Furthermore, the catalyst formed (see below)
is almost completely efficient in regard to the use of H
2
O
2
for oxidation of
substrates [118].
Recent Mechanistic Insights Until recently, most attention focused either on the
coordination chemistry of the Mn-tmtacn family of complexes or on their appli-
cation in functional group transformation, such as the oxidation of alkenes. A key
challenge faced was probing the molecular nature of the catalysts under the reaction
conditions. In part this was a consequence of their high activity and hence the low
catalyst loadings employed. The complex coordination and redox chemistry, not
least ligand exchange and disproportionation reactions, complicates unraveling the
mode of action of this versatile oxidation catalyst. By analogy with manganese
porphyrin and permanganate chemistry, the obvious candidates for the active
catalytic species are high-valent manganese-oxo species as well as radical inter-
mediates (the latter being readily excluded by the retention of stereochemistry
observed during catalysis). The first concerted efforts to explore the mechanistic
aspects of the catalysis were reported by Hage and coworkers [119], who identified
the formation of carboxylic acid-bridged dinuclear manganese complexes upon
reduction of 6 in aqueous media and by Lindsay-Smith and coworkers in their
studies on the oxidation of cinnamates in buffered aqueous media, primarily using
mass spectrometry [94i,k, 120, 121, 122].
A key feat ure of catalysis with complex 6 was the frequent observation of an
induction period prior to initiation of su bstrate conversion. This indicated that the
original [Mn
2
O
3
(tmtacn)
2
](PF
6
)
2
complex (6)hasfirst to be converted to a catalyt-
ically active species. Indeed , it was found that the catalytic activity of Mn-tmtacn was
significantly increased when it was pre-treated with excess of H
2
O
2
prior to the
addition of the substrate (in the case of benzyl alcohol oxidation) [94c]. From the
16-line spectrum obtained by electron paramagnetic resonance spectroscopy
(EPR) measurements it was inferred that the Mn
IV
–Mn
IV
dimer was reduced by
H
2
O
2
to a dinuclear Mn
III
–Mn
IV
mixed-valent species in acetone. The intensity of
the mixed-valent species gradually diminished, with the subse quent appearance of
an Mn
II
species. EPR stud ies of the catalysts under comparable cat alytic oxidation
conditions using alkenes as subst rates instead of alcohols showed again the mixed-
valence Mn
III
–Mn
IV
dimer [40a, 97]. B ased on EPR data, similar manganese species
were identified during related phenol oxidation experiments [120]. Barton et al.
proposed the formation of an Mn
V
¼ O intermediate during the oxidation of 2,6-di-
tert-butylphenol with Mn-tmtacn and H
2
O
2
[121]. In electrospray mass spectro-
metry (ESI/MS) experiments the mononuclear Mn
V
¼ O species could indeed be
assigned [122]. This species was also generated in oxidation reactions using a
mononuclear Mn
IV
complex and from an in situ prepared Mn
II
complex using
MnSO
4
and free tmtacn ligand [94g]. A key question arises, however, as to th e
relevance of these species. It was subsequently shown that the features observed by
EPR spectros copy were only in part related to th e catalysis its elf but w ere mostly
11.4 Epoxidation and cis-Dihydroxylation of Alkenes
j
399
related to disproportionation reactions at the high catalyst concentrations em-
ployed in mechanistic studies [118]. Furthermore, it was not demonstrated that the
Mn
v
¼ O specie s observed by ESI-MS were active in the catalytic oxidation of
alkenes.
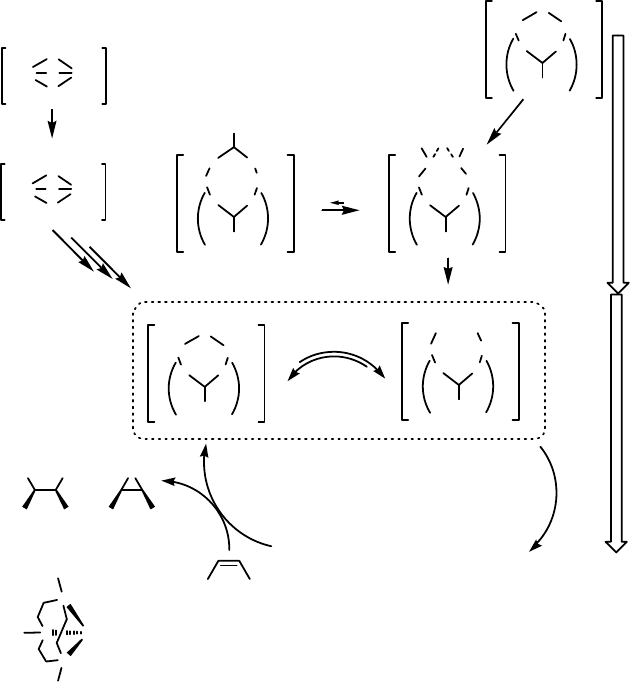
For the combination of 6 and alkyl and aromatic carboxylic acids, the high activity
and selectivity was found to be due to the in situ formation of bis-m-carboxylato-
bridged dinuclear manganese(III,III) complexes [117, 118]. These complexes were
formed through a series of redox processes (Scheme 11.15) in which H
2
O
2
acts as a
terminal reductant of the dimeric Mn
IV
complex 6. The ability of different carboxylate
ligands to tune the activity of the catalyst was found to be dependent on both the
electron-withdrawing nature of the ligand and on steric effects; that is, bulky electron-
deficient ligands gave the highest activity. By contrast, the cis-diol/epoxide selectivity
is determined primarily by steric factors; that is, more bulky carboxylic acids lead to
a higher ratio in favor of the cis-diol product. A mechanistic study of the roles played
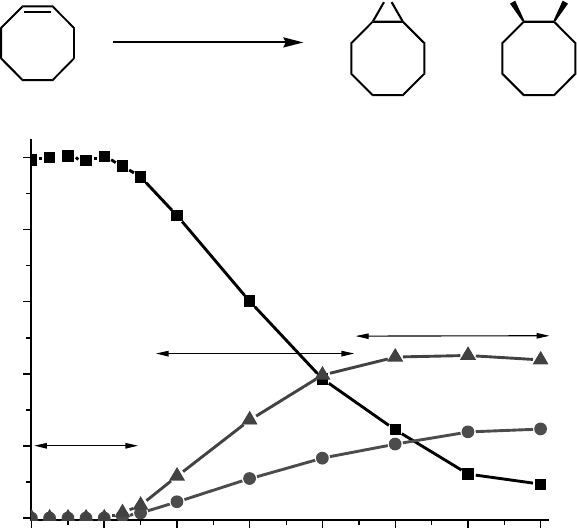
76543210
0
200
400
600
800
1000
Phase II
Phase I
t.o.n.
time (h)
Phase III
0.1 mol% [Mn
2
O
3
(tmtacn)
2
](PF
6
)
2
0.1 mol% CCl
3
CO
2
H
H
2
O
2
(aq. 50% aq.)
CH
3
CN, 0 °C
+
OHHOO
Scheme 11.15 cis-Dihydroxylation and
epoxidation of alkenes by 6: the multiple roles of
carboxylic acids in forming the active catalysts
for homogenous alkene oxidation from the
catalyst 6 allow for the effect of variations in the
composition of the reaction mixture on catalytic
activity to be understood at a molecular level.
400
j
11 Manganese-Catalyzed Oxidation with Hydrogen Peroxide
by solvent, initial catalyst oxidation state, water, carboxylic acid concentration, and
the nature of the carboxylic acid employed in determining both the activity and the
selectivity observed was reported [118]. The resting state of the active form of the
catalyst, that is, [Mn
III
2
O(m-O)(m-RCO
2
)
2
(tmtacn)
2
]
2 þ
, was identified through a com-
bination of speciation analysis employing a range of spectroscopic techniques and
isotope-labeling studies. These [Mn
III
2
(m-O)(m-RCO
2
)
2
(tmtacn)
2
]
2 þ
complexes show
coordination chemistry dependent on redox state and solvent (Scheme 11.15), but the
available evidence indicates that redox changes or change in the dinuclear structure
do not occur during catalysis.
Mn
OO
Mn
O
CCl
3
LL
III III
2
2+
Mn
OO
Mn
H
O
CCl
3
L
L
II II
2
+
Mn
OO
Mn
O
CCl
3
LL
II II
2
+
O
H HH
Mn
OO
Mn
HO
CCl
3
LL
III III
2
2+
OH
Mn
O
OMn
O
LL
IV IV
2+
Mn
OO
Mn
O
CCl
3
LL
II II
2
+
O
CCl
3
Mn
O
OMn
H
O
LL
IV IV
3+
OHHO O
or
+ H
+
+ H
2
O
+ 2 H
2
O
+ CCl
3
CO
2
H
H
2
O
2
+ H
2
O
2
+ 2 CCl
3
CO
2
H
N
N
N
Mn
= LMn
[H
2
O
2
activated complex]
+ H
2
O
- H
2
O
H
2
O
2
Phase I
P
h
a
s
e
I
I
/
I
I
I
Scheme 11.15 (Continued).
11.4 Epoxidation and cis-Dihydroxylation of Alkenes
j
401
The report by de Boer et al. [118] does not exclude the possibility of the transient
formation of high-valent mononuclear species in the catalytic cycle as proposed by
several groups [94i,k, 120, 121, 122]. However, the data available up to now support
an alternative view of the role of the tmtacn-based manganese catalysts as Lewis acid
activators of H
2
O
2
, which holds considerable implications for the study of related
homogeneous manganese oxidation catalysts and in building a conceptual bridge
with related biological systems such as dinuclear catalase and arginase enzymes (see
above) [10].
An important question, however, is whether all systems based on the manganese-
tmtacn catalyst system operate via a similar mechanism, that is, the in situ formation
of [Mn
III
2
O(m-O)(m-RCO
2
)
2
(tmtacn)
2
]
2 þ
complexes. The recent spectroscopic iden-
tification of carboxylate-bridged complexes as active catalysts does not necessarily
mean that the first systems based on 6 in which oxalic acid or ascorbic acid were used
by De Vos, Berkessel, and coworkers to promote oxidation catalysis involve the same
general mechanism [94a,b,d]. Recently de Boer et al. compared the catalytic activity of
[Mn
IV
2
(m-O)
3
(tmtacn)
2
]
2 þ
using salicylic acid, ascorbic acid, and oxalic acid as
additives by observing the spectroscopic changes during the course of the catalyzed
reactions [123]. In the case of salicyclic acid, the electronic absorption spectra of the
reaction mixture were quite different from what is observed using other carboxylic
acids. Furthermore, a mononuclear complex was isolated in which the salicylato
dianion is bound as a chelate. However, it was demonstrated through other
spectroscopic and electrochemical techniques that these differences were not
directly relevant to the catalysis itself. It was found that the role of the salicylic acid
in the catalysis, that is, to act as a carboxylato ligand for binuclear complexes, was the
same as that for other acids despite the presence of a potentially chelating hydroxyl
group.
In the case of ascorbic acid and oxalic acid as additives, their redox activity adds
an additional dimension to the catalysis they promote with 6 [123]. For both these
acids the most notable observation was the absence of an induction period; that is,
catalysis commenced immediately upon addition of H
2
O
2
. This is not surprising as
both oxalic acid and ascorbic acid are reductants. In the case of ascorbic acid, although
a [Mn
III
2
O(m-O)(m-RCO
2
)
2
(tmtacn)
2
]
2 þ
-type complex could be identified by UV-Vis
spectroscopy, EPR spectroscopy indicated that Mn
III,IV
2
dinuclear species are present
in the reaction mixture during catalysis also. Hence, although in principle ascorbic
acid may promote the system both by serving as a reductant of [Mn
IV
2
(m-O)
3
(tmtacn)
2
]
2 þ
as well as generating [Mn
III
2
O(m-O)(m-RCO
2
)
2
(tmtacn)
2
]
2 þ
-type
complexes, it is also possible that a distinct mechanism is in operation. This is
especially the case considering the systems ability to oxidize electron-deficient
alkenes.
In the case of oxalic acid an even more complex picture emerged [123]. Initially
only epoxidation is observed. However, after a certain period of time, cis-dihydrox-
ylation begins. This is in stark contrast to the other carboxylic acid-based systems
where epoxidation and cis-dihydroxylation proceed concurrently. Furthermore, the
reaction mixture does not contain EPR-active species at 77 K, and the UV-vis
absorption spectra are unstructured. Concurrently with the initiation of cis-dihy-
402
j
11 Manganese-Catalyzed Oxidation with Hydrogen Peroxide