Venables J. Introduction to Surface and Thin Film Processes
Подождите немного. Документ загружается.

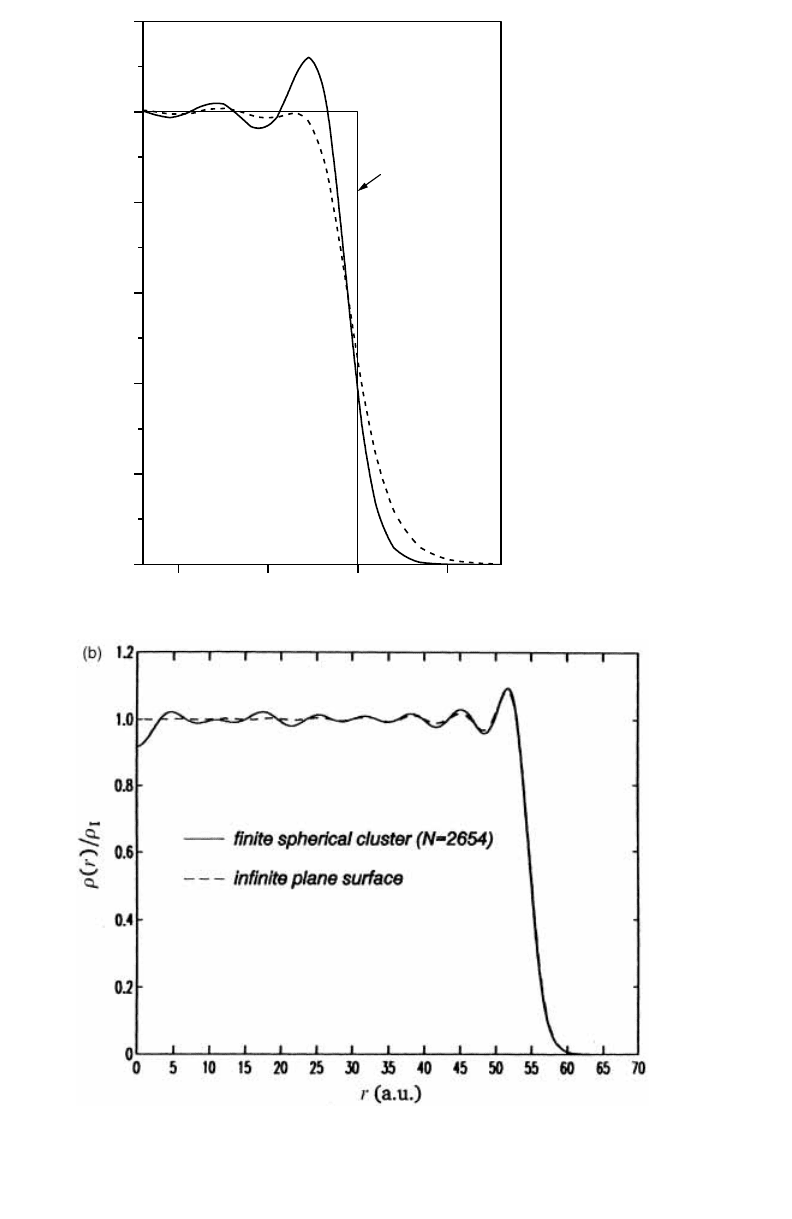
Figure 6.2. Electron density at a metal surface in the jellium model: (a) Lang & Kohn (1970)
for r
s
52 and 5; (b) comparison between a spherical cluster of 2654 simulated Na atoms
(r
s
53.96) and a planar surface for r
s
54 (after Genzken & Brack 1991, and Brack 1993,
reproduced with permission).
–1.0 –0.5 0.0 0.5
0.0
0.2
0.4
0.6
0.8
1.0
1.2
(a)
Positive
background,
ρ
+
ρ
–
for
r
s
= 2
ρ
–
for
r
s
= 5
Distance
z
(Fermi wavelengths)
Charge density (relative to bulk)
(or dip) varies with electron energy and is dominated by the highest occupied states
which vary with the exact cluster size, whereas the oscillations close to the surface are
independent of such details.
The oscillations in the electron density are called Friedel oscillations; these occur
when a more or less localized change in the positive charge density (the discontinuity
at the jellium model surface being an extreme case) is coupled with a sharp Fermi
surface. In other words, they are a feature of defects in metals in general, not just sur-
faces, and are an expression of Lindhard screening, which is screening in the high elec-
tron density limit. Screening in metals is so effective that there are ripples in the
response, corresponding to overscreening.
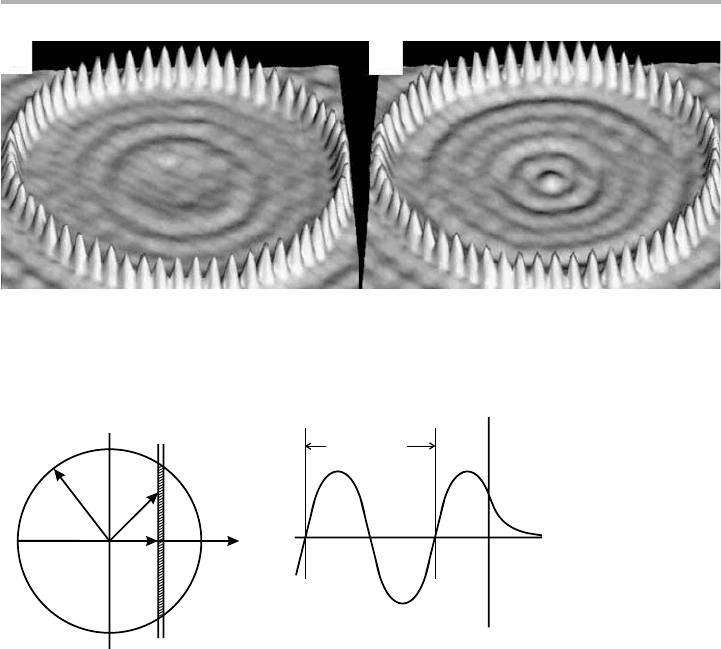
Recently, these electron density oscillations have been seen dramatically in STM
images both of surface steps, and of individual adsorbed atoms on surfaces, reported
in several papers from Eigler’s IBM group. By assembling adatoms at low temperature
into particular shapes, these ‘quantum corrals’ can produce stationary waves of elec-
tron density on the surface which are sampled by the STM tip, and the corresponding
Friedel oscillations are energy dependent; two examples from a circular assembly of 60
Fe atoms on Cu(001) are shown in figure 6.4.
Whether or not these effects can be explained in detail as yet (Fe and Cu are both
188 6 Electronic structure and emission processes
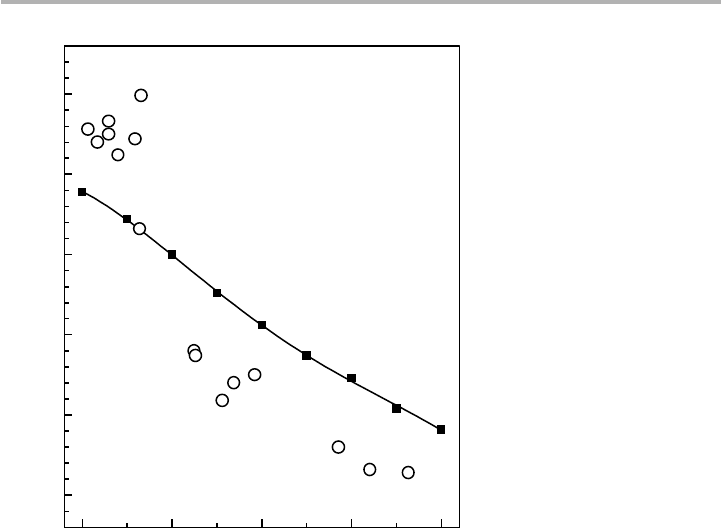
Figure 6.3. Work functions in the jellium model (full squares, Lang & Kohn 1971), compared
with experimental data for polycrystalline alkali and alkaline earth metals (open circles:
Michaelson 1977). The elements plotted are after Lang (1973) and the solid line fourth-order
polynomial fit to these points has been added.
23456
2.0
2.5
3.0
3.5
4.0
4.5
Cs
Rb
K
Na
Ba
Sr
Ca
Li
Mg
Pb
In
Cd
Hg
Zn
Ga
Al
Work function,
(eV)
radius,
r
s
(a.u.)
f
transition metals with important d-bands), these oscillations are present in free elec-
tron theory. To see how such effects arise, one needs to do as simple a calculation as
possible, and try to understand how the physics interacts with the mathematics. The
calculation done by Lang & Kohn goes roughly as follows, using figure 6.5 as a guide.
Consider pairs of states, ordered by their k-vector perpendicular to the surface, k
and 2k. Their wavefunction is
c
⬃
c
k
(z) exp i(k
x
x1k
y
y), and when 6k are combined
to vanish in the vacuum (outside the surface),
c
k
(z)⬃sin(kz -
g
F
), where
g
F
is a phase
factor, dependent on k
F
, since the origin doesn’t have to be exactly at z50. Draw a
Fermi sphere, radius k
F
, with the k-axis (perpendicular to the surface) as a unique axis,
as in figure 6.5. Make a slice at k,dk thick; the density of states g(k) is just the area of
this slice which is p(k
F
2
2k
2
). Now we can write
r
2
5n(z)5p
22
兰
g(k)|
c
k
|
2
dk, (6.3)
where the limits of integration are 0 and k
F
, and with a bit of manipulation you should
get the result
n(z)5n¯ [113cos{2(k
F
z2
g
F
)}/(2k
F
z)
2
1O(2k
F
z)
23
], (6.4)
6.1 The electron gas 189
Figure 6.4. A ‘quantum corral’ of 60 Fe atoms assembled and viewed on Cu(001) by STM at
4K. The tip imaging parameters are (a) V
t
5110 mV and (b) 210 mV, with current I51 nA
(after Crommie et al. 1995, reproduced with permission).
(a) (b)
Figure 6.5. (a) Cross section of the free electron Fermi surface, radius k
F
; (b) the combination
of traveling wave states 6k near a surface. See text for discussion.
k
dk
x
k
z
k
F
k
z
(a)
(b)
(2k)()
–1
F

where the O-notation means ‘of order (2k
F
z)
23
’. Here n¯ is the electron density in the
bulk; the symbols n¯ and
r
2
are used interchangeably. The point which is specific to 2D
surfaces and interfaces is the dependence on (2k
F
z)
22
. For impurities or point defects,
the result is O(2k
F
z)
23
, which is due to 3D geometry. For corrals on the surface with
cylindrical geometry, we encounter various types of Bessel function, for the same
reasons as in chapter 5. In scattering/perturbation theory terms, the characteristic
length, (2k
F
)
21
, is due to scattering across the Fermi surface without change of energy.
The same length occurs in the theory of superconductivity and charge density waves;
these features can be explored further via problem 6.1.
It is interesting that the jellium model also gives, though not so impressively, values
for the surface energy of the same metals as shown later in figure 6.10. The agreement
is again excellent for the heavier alkali metals, but fails dramatically for small r
s
. This
arises from the need to include the discreteness of the positive charge distribution asso-
ciated with the ions, a point which was recognized in Lang & Kohn’s original paper.
With a suitable choice of pseudopotential, agreement is much improved (Perdew et al.
1990, Kiejna 1999).
6.1.2 Beyond free electrons: work function, surface structure and energy
There have been many developments since Lang & Kohn to extend this approach, first
to s-p bonded metals and then to the complications of transition metals involving d-
electrons, and in the case of the rare earths, f-electrons as well. The d-electrons give an
angular character to the bonding, often resulting in structures which are not close-
packed, e.g. b.c.c. (Fe, Mo, W, etc.) or complex structures like
a
-Mn. This is in contrast
to s-p bonded metals which typically are either f.c.c. or h.c.p. There are many chal-
lenges left for models of metallic surfaces.
To start we need a few names of the methods, for example ‘nearly-free electron’
method, pseudopotentials, orthogonalized plane waves (OPW), augmented plane
waves (APW), Korringa–Kohn–Rostoker (KKR), tight-binding, etc. These long-
standing methods are described by Ashcroft & Mermin (1976). For surfaces, an intro-
ductory account of electronic structure is given by Zangwill (1988), which contrasts
with a highly detailed version from Desjonquères & Spanjaard (1996). Typically tight-
binding (where interatomic overlap integrals are thought of as small) is taken as the
opposite extreme to the nearly free electron model (where Fourier coefficients of the
lattice potential are thought of as small). However, this is more apparent than real, in
that both pictures can work for arbitrarily large overlap integrals or lattice potentials;
the only requirement is that the basis sets are complete for the problem being studied.
This of course can lead to some semantic problems: methods which sound different
may not in fact be so different; in particular, when additional effects are included they
are almost certainly not simply additive.
The basic feature caused by including the ions via any of these methods is that the
electron density near the surface is now modulated in x and y with the periodicity of
the lattice; an early calculation which shows this for the lowest atomic number metal
lithium is given in figure 6.6. So there are now two length scales in the problem which
190 6 Electronic structure and emission processes
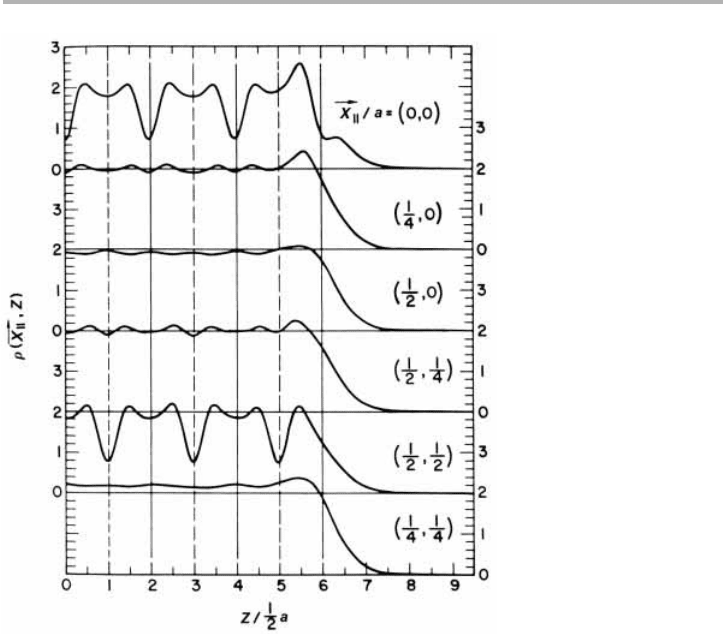
compete; surface states have oscillation periods with no simple relation to the lattice
period in the z-direction.
Contrary to the free electron starting point, it has more recently proved fruitful to
consider models based on wavefunctions relatively localized in real, rather than recip-
rocal, space, and to construct interatomic potential functions arising from atomic-like
entities interacting with the electron gas in which the ‘atom’ is embedded (Sutton 1994,
Pettifor 1995, Sutton & Balluffi 1995). The resulting methods are known as embedded
atom models (EAM) or effective medium theories (EMT); in these models the embed-
ding energy DE is expressed in terms of the cohesive function E
c
(n), as
DE5E
c
(n)1DE
c
, (6.5)
where the correction energy DE
c
differs between the various schemes, but is relatively
small.
The cohesive function E
c
(n) is a function of the homogeneous electron gas density n
in which the atom is embedded (Jacobsen et al. 1987, Jacobsen 1988, Nørskov et al.
1993). The cohesive energy, and the component E
c
(n¯) at the optimum density n¯ is
shown for the 3d transition metals in figure 6.7. A major effect of these models is to
6.1 The electron gas 191
Figure 6.6. Valence electron density at several x
//
points for Li(001) in a pseudopotential
calculation (from Alldredge & Kleinman 1974, reproduced with permission, after Appelbaum
& Hamann, 1976).
show clearly that metallic binding is strongly non-linear with coordination number.
The first ‘bonds’ to form are strong, and get progressively weaker as extra metal atoms
are added to the first coordination shell. Some of these effects were exhibited by the
experimental examples described in section 5.4. There are many subtleties in the 3d
series resulting from magnetism; here the overall cohesion peaks before and after the
middle of the transition series, unlike the 4d and 5d series, where cohesion from the d-
bands peaks in the middle. Note that this particular calculation does not include spin-
correlation effects, but some of these are discussed in relation to magnetism in section
6.3.
Many metal surface relaxations and reconstructions are due to competing electronic
and vibrational effects of a quite complex kind. For example, reconstructions of tran-
sition metals are often subtle, such as the W(001)231 and the ‘almost 231’ incom-
mensurate Mo(001) structures at low temperatures mentioned in section 1.4.3. These
structures are driven by (angular) bonding instabilities at low temperature and by
anharmonic lattice dynamics at high temperature (Inglesfield 1985, Estrup 1994,
Titmus et al. 1996). F.c.c. noble metal surfaces can be strongly affected by their d-elec-
trons, interacting with the ions and the other electrons. Although Ag(111)(131) is
unreconstructed, Au(111) has a uniaxially compressed herringbone (roughly 2331)
192 6 Electronic structure and emission processes
Figure 6.7. Calculated cohesive energies and the equilibrium radius r
s
for the 3d transition
series, comparing effective medium theory (EMT) (open circles) with KKR methods
(Morruzzi et al. 1978, closed circles). The modified EMT (open squares) corresponds to EMT
applied at the density given by the KKR method (Jacobsen et al. 1987, and Jacobsen 1988;
redrawn with permission).
K Ca Sc Ti V Cr Mn Fe Co Ni CuK Ca Sc Ti V Cr Mn Fe Co Ni Cu
2
3
4
Radius, (a.u.)
r
s
Radius,
(a.u.)r
2
0
4
6
8
Cohesive ener
gy (eV/atom)
Cohesive ener
gy (eV/atom)
EMT
Modified EMT
Moruzzi et al.
EMT
Modified EMT
Moruzzi et al.

reconstruction, Au(001) has a quasi-hexagonal surface layer giving a (roughly 2035)
diffraction pattern (Van Hove et al. 1981, Barth et al. 1990), and Pt(111) has a 737
reconstruction which can be removed by depositing Pt adatoms (Needs et al. 1991,
Bott et al. 1993). This last case shows that the surface structure and lattice parameter
of a metal can be a function of the supersaturation D
m
of its own vapor, and that
adatoms and surface reconstructions can change the surface stress. This possibility is
also well known from adsorption studies of rare gases on graphite, as discussed in
section 4.4.
An increasing number of theorists are sufficiently practical and public-spirited that
they collaborate closely with experimentalists, and make their computer codes avail-
able to others for work on specific problems. It is a welcome recent development that
theorists have addressed the problem of ‘understanding’. By this I mean that they
acknowledge that the ‘true’ solution is obtained by keeping all the terms in the
Schrödinger equation that they can think of, but that this doesn’t necessarily help one
understand trends in behavior, or help one make predictions. Pettifor (1995), for
example, starts with a quote from Einstein: ‘As simple as possible, but not simpler’.
This is excellent: with such an attitude there is real prospect that we can ‘understand’
a higher proportion of theoretical models than we would be able to otherwise.
Increasingly what counts is the speed of the computer code; if this speed scales with
a lower power of the number, N, of electrons in the system, then more complex/larger
problems can be tackled; O(N) methods are in! For example, because the interactions
between atoms and the electron gas are parameterized initially, EMT calculations are
fast enough that they can be used to simulate dynamic processes such as adsorption,
nucleation or melting on metal surfaces; here an approximate electronic structure cal-
culation is being done for each set of positions of the nuclei, i.e. at each time step
(Jacobsen et al. 1987; Stolze 1994, 1997). This requires computer speeds that would
have been inconceivable just a few years ago. It is now feasible to download EMT pro-
grams from a website in Denmark (see Appendix D) in order to run them for a class
project in Arizona! There are real possibilities for experiment–theory collaborations
here which were impractical just a few years ago.
6.1.3 Values of the work function
There are several methods of measuring the work function, as described by Woodruff
& Delchar (1986, 1994), by Swanson & Davis (1985) and by Hölzl & Schulte (1979)
amongst others. The work function varies with the surface face exposed, as shown for
several elemental solids in table 6.2. Note that for b.c.c. metals, the surfaces decrease
in roughness in the order (111), (100), (110) presented, whereas for f.c.c. the same order
corresponds to an increase in roughness. These variations are responsible for several
interesting effects, as described here and in the next section.
A polycrystalline material, with different faces exposed, gives rise to fields outside the
surface, referred to as patch fields. Such fields are very important for low energy elec-
trons or ions in vacuum, and can thereby influence measurement accuracy in surface
experiments. Molybdenum is often used for such critical parts of UHV apparatus,
6.1 The electron gas 193

194 6 Electronic structure and emission processes
Table 6.2. Experimental work functions for metals assembled by Michaelson (1977),
compared with calculations by Perdew, Tran & Smith (1990; PTS), Skriver &
Rosengaard (1992; SR) and Methfessel, Hennig & Scheffler (1992; MHS), plus
others as indicated in the last column
Model
Metal/ Face Experiment* Model Model (MHS)
structure {hkl} (eV) (PTS) (SR) 1 others*
Li 111 2.90
b.c.c. 100 2.9 (poly) 2.92 3.15 3.03
b
110 3.09 3.33 3.27
b
Na 111 2.54
b.c.c. 100 2.75 (poly) 2.58 2.76 2.66
b
110 2.75 2.94 2.88
b
K 111 2.17
b.c.c. 100 2.30 (poly) 2.21 2.34 2.27
b
110 2.37 2.38 2.44
b
Cs 111 1.97
b.c.c. 100 2.14 (poly) 2.01 2.03 2.04
b
110 2.17 2.09 2.19
b
Al 110 4.06 3.81
f.c.c. 100 4.4160.03
a
3.62 4.50
a
111 4.24 3.72 4.54 4.09
b
Cu 110 4.48 4.48
f.c.c. 100 4.59 5.26
111 4.98 5.30
Ag 110 4.52 4.40 4.23
f.c.c. 100 4.64 5.02 4.43
111 4.74 5.01 4.67
Au 110 5.37 5.40
f.c.c. 100 5.47 6.16
111 5.31 6.01
Nb 111 4.36
b.c.c. 100 4.02 3.68
110 4.87 4.80 4.66
Mo 111 4.55
b.c.c. 100 4.53 4.49
110 4.95 5.34 4.98
W 111 4.47
b.c.c. 100 4.63
110 5.25 5.62
Note: *Error bars and other calculations by: (a) Inglesfield & Benesh (1988); (b) Perdew
(1995).

because the work function doesn’t vary more than 0.4 V between the low index faces
(table 6.2), whereas Nb and W, which are otherwise similar, have variations of around
0.8 V.
The origin of this face-specific nature of the work function can be seen qualitatively
by considering jellium again. First, we can see from figures 6.1 and 6.2 that the nega-
tive charge spills over into the vacuum, causing a dipole layer, whose dipole moment is
directed into the metal. Now we use Gauss’ law and show that
DV(volts)5
s
d/
«
0
5pN/
«
0
, (6.6)
where the sheets of charge, surface charge density
s
, are separated by a distance d.To
get an idea of how big the potential change is, think of each atom on the surface
(Nm
22
) having a charge of 1 electron separated by 0.1 nm (1 ångström). With N523
10
19
m
22
, p51.6310
229
Cm, and
«
0
58.854310
212
Fm
21
, we get DV536.14 V. This
value is perhaps 2–5 times as large as most voltage (energy) differences between the
vacuum level and the bottom of the valence band (which is also the conduction band
in monovalent metals).
So a charge separation of ,0.5Å is needed to produce the desired effect. Is it a coin-
cidence that this is the same order of magnitude as the Bohr radius, a
0
50.0529 nm?
Not really: the reasons for both effects, the spill over of electrons due to the need to
reduce kinetic energy, are the same! This is, of course, a zero order argument: to get the
numbers right we have to go back to exchange and correlation energies, and the details.
However, models may contain rather arbitrary parameters. For example, the ‘corruga-
tion factor’ introduced into the ‘structureless pseudopotential model’ (Perdew et al.
1990) sounds rather dubious, although it moves the model in the right direction
(Perdew 1995). Brodie (1995) has proposed a model, building on the idea of corruga-
tion, which is ‘too simple’ in Einstein’s sense; this model should be ignored since it is
incapable of further elaboration.
While on this subject, we can note the unit to describe dipole moments, the Debye
(D). This is 10
218
esu·cm53.33310
230
Cm. Thus 1 electron charge31Å54.81 D.
Adsorbed atoms change the work function considerably, but only alkalis give rise to
dipole moments this large; for example Cs adsorbed on W(110) at low coverages has
been calculated to have a dipole moment of at least 9D (see e.g. table 2.2. in Hölzl and
Schulte 1979); in this case the single electron charge distribution would be shifted by
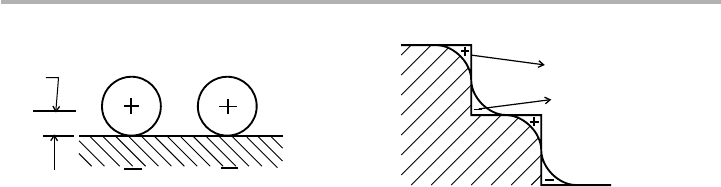
about d50.2 nm. This simple picture is illustrated in figure 6.8(a), and corresponds to
(partial) ionization of the alkali, a model first introduced by Langmuir in 1932 and
developed by Gurney in 1935. But we need to be careful about inclusion of the image
charge, and the nature of bonding, which varies with coverage and is the subject of
ongoing discussion (Diehl & McGrath 1997).
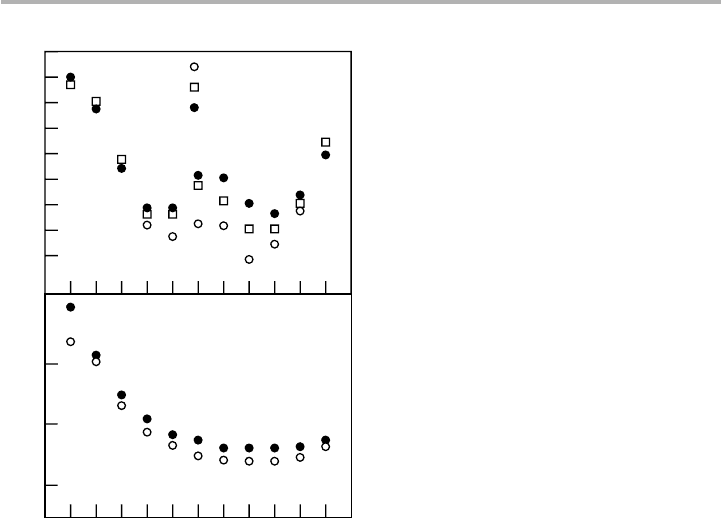
The same arguments about electron spillover tell us that stepped, or rough surfaces
will have lower work function than smooth surfaces, due to dipoles associated with
steps, pointing in the opposite direction to the dipole previously considered for the flat
surface. A schematic (top view) of this situation, referred to sometimes as the
Smoluchowski effect after a 1941 paper, is shown in figure 6.8(b). Experiments on
vicinal surfaces, close to low index terraces, do indeed show that the work function
6.1 The electron gas 195
decreases linearly with step density, as shown in figure 6.9; this implies that there is a
well defined dipole moment per ledge atom, around 0.3 D for steps parallel to [001] on
W(110) and varying with step direction on Au and Pt(111) surfaces (Krahl-Urban et
al. 1977; Besocke et al. 1977; Wagner 1979).
Fascinatingly, work function changes as a function of temperature can be used to
define 2D solid–gas phase changes via these same effects. An adatom has a dipole
moment which depends both on its chemical nature and on its environment. In a solid
ML island the dipole moment per atom is considerably smaller than in the 2D gas. This
effect has been used to map out the gas–solid phase boundary for Au/W(110) at high
temperatures (Kolaczkiewicz & Bauer 1984). Phase changes in adsorbed layers are dis-
cussed in more detail in chapter 4.
6.1.4 Values of the surface energy
While the work function is a sensitive test of electronic structure models, particularly
exchange and correlation, surface energies are sensitive tests of our understanding of
cohesion. Surface energies clearly increase, in general, with sublimation energies, and
this has led to many studies trying to embody such relationships into universal poten-
tial curves, or into other semi-empirical models (Rose et al. 1983, deBoer et al. 1988).
But the effective medium and density functional models have proven to be more
durable, and we may well now have arrived at the point where these models are more
accurate than the experiments, which were almost all done a long time ago on poly-
crystalline samples, sometimes under uncertain vacuum conditions. An example of a
microscope based sublimation experiment for Ag, which has stood the test of time, and
which agrees with recent calculations within error, is that by Sambles et al. (1970).
Some experimental and theoretical values for a range of metals are given in table 6.3.
As in the case of the work function, the starting point for models of alkali and other
s-p bonded metals has been the jellium model. As was shown in the original Lang &
Kohn papers, jellium has an instability at small r
s
values, which is due to the neglect of
the ion cores. This topic has been pursued by Perdew et al. (1990), Perdew (1995) and
Kiejna (1999), who have been interested in exploring the simplest feasible pseudopo-
tential models for such metals, and in particular obtaining trends in calculations as a
196 6 Electronic structure and emission processes
Figure 6.8. Origins of face- and adsorbate-specific work function: (a) dipoles due to charge
transfer from adsorbates; (b) top-view of a stepped surface showing smoothing of the charge
distribution around the steps (after Gomer 1961, and Woodruff & Delchar 1986, redrawn with
permission).
(a) (b)
d
image chargesimage charges
+ve
-ve
charge
s
charge
s