Marshall L. Stoller, Maxwell V. Meng-Urinary Stone Disease
Подождите немного. Документ загружается.

134 Chin
tibiotics, are excreted in large concentrations in the urine, the calculated UAG will often
be positive. In addition, other urinary electrolytes such as calcium and magnesium are
generally not part of the calculation, although technically they do make up a small part
of the cation content, and excretion of these ions can be quite variable. Nonetheless, the
calculated UAG, although best viewed as a semiquantitative tool, is clinically useful in
the evaluation of renal-mediated metabolic acidosis.
U
RINARY OSMOLAR GAP
A second method of estimating NAE involves calculation of a urine osmolar gap
(UOG). Because the urine osmolarity should be the sum of the major charged particles
and the major uncharged particles, the following should hold true: urine osmolarity =
(positive charged) + (negative charged) + (uncharged). We can assume the number of
positively charged particles to be equal to the number of negatively charged ones be-
cause urine is not a charged substance, and the major uncharged particles are urea and
glucose. This brings the relationship to: urine osmolarity = 2(Na
+
+ K
+
+ NH
4
+
) + urea
+glucose (in mmol/L). Solving for ammonium results in: NH
4
+
= 0.5 {urine osmolarity
– [2(Na
+
+ K
+
) – urea – glucose]} (in mmol/L). If the daily urinary volume is known, 24-
h ammonium excretion can be estimated. The correlation of this method of urinary
ammonium estimation to measured urinary ammonium is quite good, and may be more
precise and accurate than the UAG (37).
The limitations of using the UOG are similar to that of using the UAG; if there are
other unmeasured substances, the calculation may inaccurately predict the urine ammo-
nium content. In using the UOG, other osmotically active substances such as glucose,
mannitol, or glycerol must not be present in large quantities.
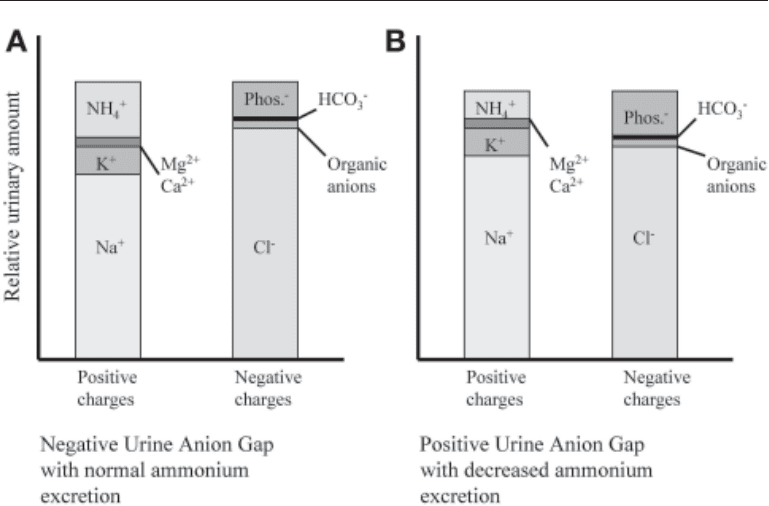
Fig. 7. Urine anion gap with (A) normal ammonium excretion and (B) decreased ammonium
excretion.

Chapter 8 / Renal Acid–Base Balance 135
Additional Tests
Evaluation of renal tubular acidification sometimes involves additional provocative
tests to challenge the function of tubular segments. These include bicarbonate loading,
acid loading, urine-to-blood carbon dioxide gradient determination, and urine pH mea-
surement after furosemide challenge (Table 1). The table is meant only as a guideline and
to explain the rationale behind each test.
The bicarbonate-loading test evaluates proximal tubular reabsorption capability. If
significant bicarbonaturia occurs when the serum bicarbonate is brought closer to nor-
mal levels, proximal RTA is diagnosed. It should be noted that complete correction or
over correction of the metabolic acidosis with sodium bicarbonate will lead to increased
bicarbonaturia, regardless of the underlying condition, and some investigators believe
that evaluation for proximal RTA is better when plasma bicarbonate levels are kept
below normal (38). Proximal RTA usually results in fractional excretion of bicarbonate
>15% with this challenge, as well as an increase in urine pH levels from <5.5 to >6.0.
When bicarbonate loading is performed, measuring the urine-to-blood partial pres-
sure of carbon dioxide (U-B pCO
2
) gradient may be helpful in evaluating the function
of H
+
secretion in the collecting tubule. Essentially, bicarbonaturia is induced with a rise
in the urine pH >7.5. This ensures that there is enough bicarbonate in the distal segments
to bind most of the secreted protons. If proton delivery is intact, there should be a rise
in urine CO
2
levels (H
+
+ HCO
3
–
↔ H
2
O + CO
2
). The simultaneous blood pCO
2
value
is measured and subtracted from the urinary value, giving a gradient expressed in units
of mmHg. The normal value is >20 mmHg gradient. The value should be interpreted only
when the urine pH is elevated to >7.5 where urinary bicarbonate is elevated.
The results of acid loading determine whether or not there is a defect in distal acidi-
fication. In many instances, the underlying RTA disorder already gives an acidemia,
with serum pH of <7.35 and a significant metabolic acidosis. In such a circumstance, the
use of an acid stress test is probably unnecessary. In addition, the formulation commonly
causes gastrointestinal upset. Nonetheless, in cases where the diagnosis is unclear, where
there is suspected distal acidification defects in addition to a known proximal defect, or
where there is a suspected incomplete RTA, the use of an acid load can be very informa-
tive. Acid loading is usually accomplished by administering oral NH
4
Cl, 0.1 g/kg body
weight per day in divided doses (39), given orally for 3 d. Numerous variations of this
protocol are available, including shorter ones for children (40), but the goals are the
same: to induce a degree of acidemia and acidosis large enough to evaluate the appro-
priateness of acid excretion. This, in effect, tests the entire NAE process, including the
availability of ammonia, the distal ability to secrete H
+
, and the membrane integrity. Of
course, if the result of the NH
4
Cl test is abnormal, other tests may be necessary to
determine the exact mechanism of decreased NAE.
The normal response to a noncarbonic acid load is increased NAE. Urine pH should
be <5.5 and the urine titrateable acidity and ammonium content should be increased.
Because the latter two are difficult or inconvenient to measure, the urine anion gap or
urine osmolar gap may be used to estimate ammonium excretion. After NH
4
Cl loading,
a serum pH of <7.35 and a decrease in bicarbonate of >3 meq/L general indicates an
adequate load was given. Urine pH and the UAG should be measured at regular intervals,
often hourly, after an acidemia and acidosis are clearly established. A persistent urine pH
of >5.5 indicates abnormal distal acidification. Usually, NAE is also decreased. A pos-
sible exception is in one form of incomplete RTA, where an exaggerated proximal
ammonium production is felt to be the underlying defect (see section on incomplete
136 Chin
136
Table 1
Tests of Renal Tubular Function
Test Goal Method Result
Bicarbonate loading • To determine defect in bicarbonate • Sodium bicarbonate usually given • Fractional excretion of bicarbonate
reclamation orally to correct bicarbonate to
>15% in proximal RTA
near-normal levels
Urine to blood partial • To determine if there is a distal • With bicarbonate loading, ensure • Gradient (urine–blood) is normally
pressure of CO
2
acidification defect urine pH is > 7.0–7.5 and measure >20 mmHg. Abnormal value of <20
gradient measurement simultaneous blood and urine pCO
2
mmHg found in classic distal RTA,
some forms of hyperkalemic, rate-
dependent and incomplete RTA
NH
4
Cl loading • To determine if there is a distal • Oral NH
4
Cl loading of 0.1 g/kg/d • Failure to acidify urine (pH >5.5)
acidification defect
for 1–3 d and measuring serum and with inadequate ammonium for-
urine pH; consider urinary
mation suggests distal acidification
ammonium or urine anion gap
defect. Usually abnormal in distal
measures RTAs. Incomplete RTA may give pH
>5.5 with normal ammonium
Furosemide testing • To determine if there is a distal • Furosemide is given at 40–80 mg • Failure to acidify urine (pH >5.5)
acidification defect owing to orally, or an as intravenous dose;
when sodium delivery is increased
inadequate sodium delivery measure urine pH when there is a
suggests a secretory defect in the
documented naturesis
distal tubule. Usually normal in
proximal RTA, some rate-dependent
RTA, type 4 RTA
RTA, renal tubular acidosis.

Chapter 8 / Renal Acid–Base Balance 137
RTA) (41). In this case, the ammonium excretion and UAG would theoretically be
appropriate for the degree of acidemia, but the urine pH remains high because of a large
amount of nonbicarbonate buffer delivery to the distal segments.
The inadequate delivery and reabsorption of sodium in the collecting tubules can lead
to an acidosis due a lesser degree of luminal electronegativity. The furosemide test
attempts to provide an adequate sodium load to the distal segment of the nephron with
subsequent measurement of urine pH to see if acidification can take place. Sometimes,
a mineralocorticoid is given before the administration of furosemide to ensure that a
deficiency in hormone is not the underlying problem as opposed to a true defect in the
distal tubular segments. Furosemide can be given orally at 40 or 80 mg as a single dose.
Other protocols give larger, intravenous doses at 1 mg/kg (42). Urinary sodium concen-
trations of >20 meq/dl indicate a good level of sodium delivery. Urine pH is then mea-
sured. The normal response is a urine pH of <5.5, which is also seen in proximal RTA,
type 4 RTA, and some dRTAs with a voltage-dependent defect. Those with a true
secretory or permeability defect usually maintain the urine pH >5.5.
RENAL TUBULAR ACIDOSIS
The term renal tubular acidosis (RTA), first coined in the 1950s, refers to a varied
collection of disorders characterized by hyperchloremic metabolic acidosis (HCMA)
initiated and maintained by a renal defect in acid–base handling. In addition, RTA should
be diagnosed only when glomerular filtration is not significantly impaired; acidosis
associated with decreased renal mass may be difficult to separate from true tubular
defects. Determining that a HCMA exists requires a basic work-up, including arterial
blood gas analysis, chemistries and thorough review of systems. The findings of: (1) a
mild to moderate acidemia; (2) a decreased pCO
2
; (3) a decreased serum HCO
3
–
; (4) a
normal serum anion gap; and (5) no obvious gastrointestinal causes for acidosis, hint that
an RTA exists. In other situations, such as in “incomplete RTA,” the acidemia and
metabolic acidosis may be mild or absent altogether. These cases require additional
testing to demonstrate a renal tubular pathology.
The pathophysiology of RTAs can be broadly divided into two defects: (1) defects in
reclamation of filtered bicarbonate and (2) defects in distal nephron acid excretion.
Although this dichotomy aids in the evaluation and treatment of RTAs, the pathologic
mechanisms oftentimes overlap with this simplified classification. The development of
more intricate assays has led to new insights into this rather complicated spectrum of
disease. The following sections on renal tubular acidosis will be divided based on the
underlying pathophysiology of the defect, and not exclusively on the laboratory find-
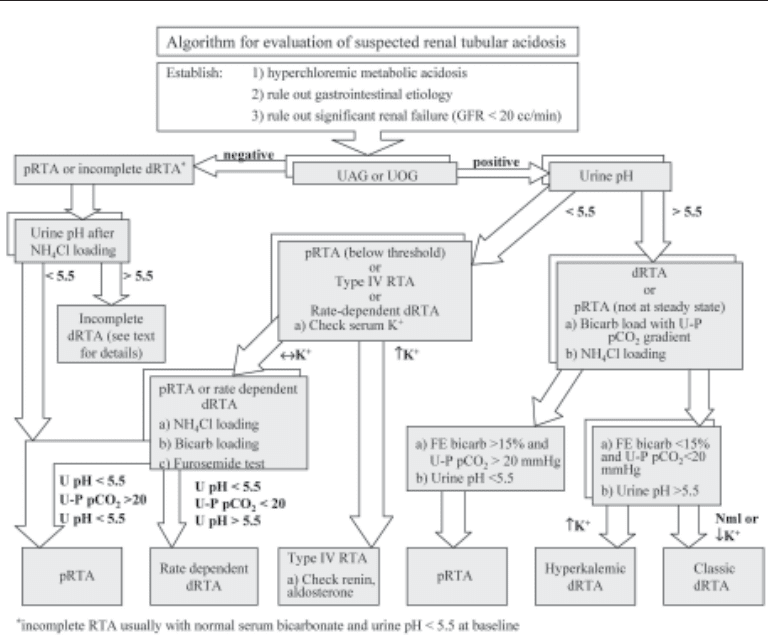
ings, such as potassium levels or urine pH. Nonetheless, a proposed algorithm based on
common laboratory tests is provided in Fig. 8.
Proximal Renal Tubular Acidosis
The proximal tubule performs two major tasks in acid–base homeostasis: the reclama-
tion of the vast majority of filtered bicarbonate and the production of ammonium neces-
sary for acid excretion in the collecting tubule. A defect in the bicarbonate reclamation
process is the primary cause of a proximal renal tubular acidosis (pRTA), also termed a
type II, RTA. This results in a lowered serum bicarbonate threshold for renal bicarbonate
wasting. When the serum bicarbonate drops below this threshold, acidification of the
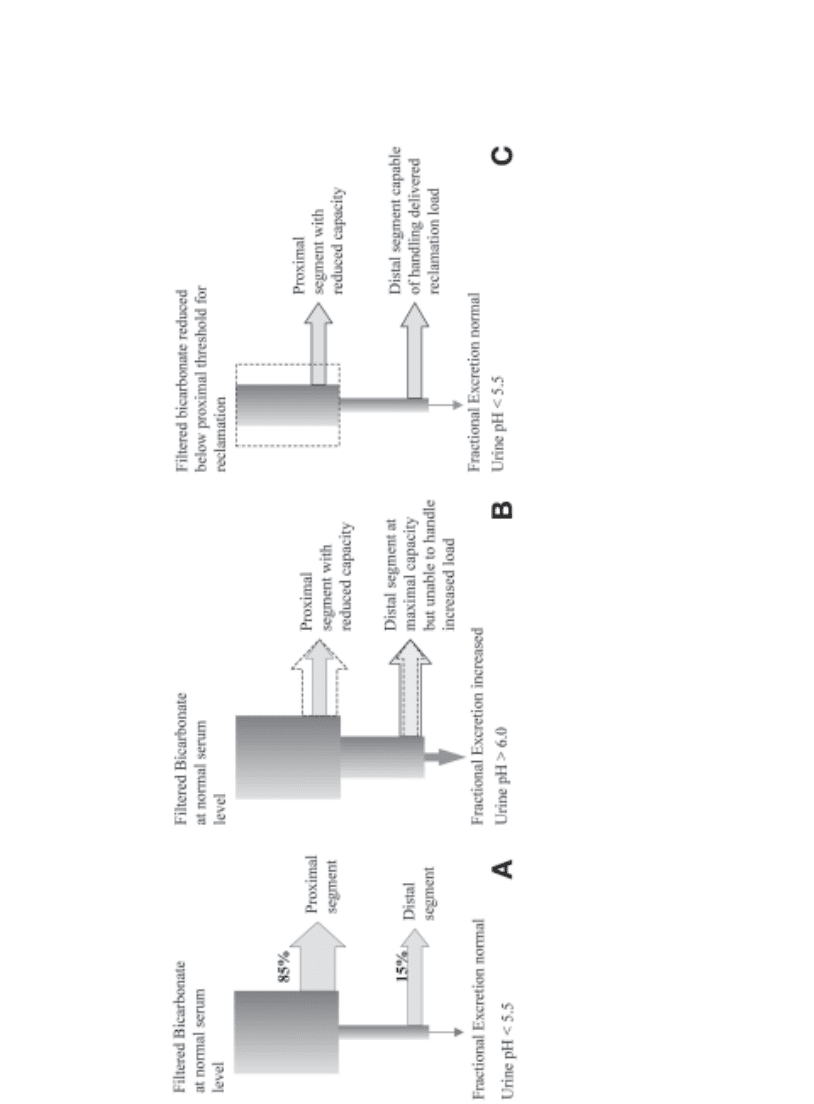
urine can be established, because there is no defect in the distal segments (Fig. 9A–C).
138 Chin
CAUSES OF PROXIMAL RTA
Proximal RTA is a rare entity. It is often seen in association with other proximal
tubular abnormalities including urinary leakage of amino acids, phosphates, glucose,
and variable amounts of protein. The combination of pRTA with other proximal tubular
problems is termed the renal Fanconi syndrome. The causes of pRTA, with or without
Fanconi syndrome, are listed in Table 2. Isolated pRTA, without renal Fanconi syn-
drome, can be found in hereditary form, both as an autosomal dominant and an autosomal
recessive type, and as a spontaneous, sporadic process (17).
As discussed in the section on proximal tubular bicarbonate handling, HCO
3
–
recla-
mation involves a Na
+
/H
+
exchanger (NHE3) as well as an H
+
-ATPase to secrete protons
into the lumen, and a basolateral Na
+
/HCO
3
–
cotransporter (kNBC1) to move intracel-
lular bicarbonate out into the capillary circulation. Mutations in these ion transporters
are the pathologic defects in some cases of isolated pRTA. In addition, carbonic anhy-
drases (CA2, isoform 2 located intracellular and CA4, isoform 4 on the luminal surface)
are critical enzymes in the function of this tubular segment. Abnormalities in CA2 have
been found in patients with a combined proximal and distal renal tubular acidosis.
Fig. 8. Differentiating the types of renal tubular acidoses.
Chapter 8 / Renal Acid–Base Balance 139
Fig. 9. (A)
Normal bicarbonate reabsorption with minimal urinary bicarbonate loss.
(B) Proximal RTA: serum bicarbonate above lowered “threshold”
for proximal reabsorption, resulting in active bicarbonaturia.
(C)
Proximal RTA: serum bicarbonate below lowered “threshold” for proximal reabsorp-
tion with cessation of bicarbonaturia.
139

140 Chin
Table 2
Classification and Causes of Proximal
Renal Tubular Acidosis (Type 2 RTA)
Primary disorder
• Isolated
°
Hereditary
Autosomal recessive
Autosomal dominant
Carbonic anhydrase II deficiency
°
Sporadic/transient
• With generalized tubular abnormalities (Fanconi syndrome)
°
Hereditary
°
Sporadic
°
Inborn errors of metabolism
Tyrosinemia
Cystinosis
Wilson’s disease
Lowe’s disease
Galactosemia
Glycogen storage disease type I
Secondary/acquired disorder
• Toxin/Drug (oftentimes with Fanconi Syndrome)
°
Heavy metals
Lead
Cadmiun
Mercury
°
Prescribed medications
Cisplatin
Gentamicin
Streptozocin
Cyclosporine
Ifosfamide
Tetracycline
Valproic acid
Acetazolamide
• Systemic diseases
°
Multiple myeloma
°
Amyloidosis
°
Light-chain disease
°
Sjogren’s syndrome
°
Hyperparathyroidism
• Others conditions
°
Renal transplantation
°
Medullary cystic kidney disease
CLINICAL PRESENTATION
Children with pRTA usually present for initial evaluation based on growth or growth
velocity retardation. In autosomal recessive types, children may also present with mental
retardation or ocular problems (38). When pRTA is found with other transport defects,
as in cystinuria or Fanconi syndrome, the presenting problems may be varied.

Chapter 8 / Renal Acid–Base Balance 141
Many autosomal recessive cases have, as the primary defect, a mutation in the gene
coding for the kNBC1 cotransporter. Because this protein is also found in corneal endo-
thelial cells (43,44), ocular findings may be presenting symptoms as well. An mRNA
splice variant of the same gene codes for a similar transporter in the brain and spinal cord,
and possibly accounts for the mental retardation seen in some cases (15).
Autosomal dominant pRTA has been found in only one described pedigree (45).
The molecular aspects have not been clarified, but there is speculation of a mutation
in the NHE3-coding gene for the Na
+
/H
+
exchanger. Sporadic cases in children are felt
to be caused by immaturity of the NHE3 exchanger (46). In infants, the proximal
tubular bicarbonate threshold is lower than that of older children or adults (46). Matu-
ration of the NHE3 exchanger may be just slower in these children identified as having
spontaneous, sporadic pRTA.
The acquired forms of pRTA, often in association with Fanconi syndrome, involve
various tubular transport abnormalities, some caused by medications and toxins. Amino
acid and phosphate transport require Na
+
-dependent transporters. Therefore, transporter
abnormalities or disruption of sodium gradients would impair absorption of many sub-
stances handled normally by the proximal tubule. Theories of membrane co-transporter
defects, Na
+
gradient defects and energy depletion problems have all been proposed (47,48).
L
ABORATORY FINDINGS AND TESTING IN PRTA
The lowered proximal tubular cell threshold for bicarbonate wasting in pRTA results
in large amounts of bicarbonate excretion when the serum bicarbonate level is above this
threshold; distal mechanisms for bicarbonate reclamation are overwhelmed by the
amount of bicarbonate coming out of the proximal segment in the bicarbonate losing
stages. Consequently, urine pH is elevated during active bicarbonaturia. Cations must
be excreted with the bicarbonate to maintain electroneutrality. As a result, sodium,
potassium and magnesium are lost during this phase. Hypokalemia is often a useful
diagnostic criterion for pRTA during bicarbonate loading tests.
Once serum bicarbonate concentrations drop below the threshold of bicarbonate rec-
lamation capacity, typically around 10–15 meq/L, bicarbonaturia returns to normal low
levels and urine pH can be acidified to <5.5, because distal acidification and NAE are
intact. Active potassium and sodium losses will cease at this point. Studies have dem-
onstrated intact sodium, phosphorous and calcium handling in patients with isolated
pRTA (49). Acid homeostasis is maintained, albeit at a lower serum bicarbonate level.
Therefore, calculations to estimate NAE, such as urinary anion gap and urinary osmolar
gap, are in the normal range assuming they are measured at steady state. Certainly if there
is a significant Fanconi syndrome, abnormal urinary levels of anionic molecules, such
as phosphates and organic anions, may make the UAG abnormal.
Confirmation of pRTA involves looking at baseline potassium levels (usually normal
at steady state in pRTA), urine pH measurement, evaluation for glucosuria, phospha-
turia, and aminoaciduria (presence or absence of Fanconi syndrome), and bicarbonate
loading. As serum bicarbonate approaches normal levels with alkali loading, finding a
fractional excretion of bicarbonate of >15% suggests a pRTA.
Additional provocative testing includes bicarbonate loading with measurement of
urine to blood partial pressure of CO
2
(U-B pCO
2
) gradient. Theoretically, if bicarbonat-
uria is present and distal delivery of H
+
is intact, there should be a build up of H
2
CO
3
within the distal nephron with some slow dehydration of H
2
CO
3
to form CO
2
. The CO
2
builds up in the urine, and the resulting gradient with blood pCO
2
levels is measured. In

142 Chin
suspected pRTA, a gradient of >20 mmHg is consistent with intact distal H
+
secretion
(38,50). Urine must be carefully collected to prevent equilibration of urine pCO
2
with
atmospheric conditions.
Acid loading in isolated pRTA should theoretically result in appropriate acid excre-
tion. Interestingly, partially impaired ammoniagenesis has been found in some indi-
viduals with pRTA. Usually, net endogenous acid excretion is normal in pRTA as
evidenced by the ability to acidify urine to pH <5.5 and to maintain a constant, although
lower, serum bicarbonate level under average dietary conditions. Laboratory findings
and results of provocative testing are summarized in Table 3.
A recent investigation suggested that under an exaggerated acid “stress” as in exogen-
ous NH
4
Cl loading, ammonium excretion was significantly less in patients with pRTA
than in controls (51). In this same study, pRTA patients at baseline relied on a higher
percentage of titrateable acid excretion than ammonium excretion when compared with
normal controls; ammoniagenesis was inappropriately low given the degree of acidemia.
Although the reasoning for this finding is unclear, it has been suggested that perhaps
the intracellular pH in the proximal tubular cells is higher than that of normal individuals
(24,51). Because bicarbonate reabsorption and ammoniagenesis would both be down
regulated if intracellular pH were elevated, this explanation would fit the laboratory
findings. In addition, citrate excretion is normal in isolated pRTA. In a chronic acidotic
state, proximal tubule reabsorption of this anion would be predictably higher than nor-
mal (because citrate is a potential bicarbonate source) with resulting hypocitraturia.
However, it is well documented that citraturia in pRTA is essentially normal at steady
state (49). An elevated intracellular pH in the proximal tubular cell could also explain
why the proximal tubular segment does not more avidly retain citrate in pRTA.
T
REATMENT OF PRTA
Alkali replacement remains the mainstay of treatment for pRTA of any cause.
Treating children with alkali gives the best chance at improving growth and avoiding
co-morbidity (50). In adults with acquired forms of pRTA, the chronic acidosis may
cause problems with protein catabolism (52–54) and bone disease (49,55). Oftentimes
adults are asymptomatic with the degree of acidosis, and there is always debate as to
how aggressively to treat adults with only moderate degrees of pRTA.
Alkali can be given in the form of a bicarbonate salt or as an organic anion salt such
as citrate. The amounts needed to correct the acidosis are usually very high, sometimes
in the range of 15–20 meq/kg for children (38). Even at these high doses, the acidosis may
not be fully corrected. The resulting bicarbonaturia will lead to increased urine cation
losses. Hypokalemia, probably caused by both the bicarbonate loss and the increased
distal tubular sodium delivery, requires replacement; a combination sodium and potas-
sium citrate salt is often appropriate. Although urinary calcium loss is not a finding of
pRTA in steady state, the increased distal tubular flows from alkaline salt replacement
can increase calciuria. In individuals with renal Fanconi syndrome, replacement of
phosphorous and amino acids may also be required. The rickets of Fanconi syndrome in
children can be improved with phosphorous and vitamin D to promote gastrointestinal
absorption of the phosphorous.
Distal Renal Tubular Acidosis
Distal tubular function is important for excretion of endogenously accumulated acid
and the acidification of urine. Distal renal tubular acidosis (dRTA) is characterized by

Chapter 8 / Renal Acid–Base Balance 143
143
Table 3
Guideline for Renal Tubular Acidosis
Classic Hyperkalemic Rate-limited Incomplete Type 4
pRTA dRTA dRTA dRTA RTA RTA
Presence of HCMA
YY YYNY
Serum K
+
↔
or ↓↔ or ↓↑ ↔ ↔↑
Urine pH (baseline) <5.5 >5.5 >5.5 <5.5 >5.5 <5.5
UAG or UOG – + + + – +
NH4 excretion ↔↓ ↓↓↑↓
Citrate excretion ↔↓ ↓↓↓↔
or
↓
Bicarbonate load with FE Bicarb >15%
YN NNNN
U-B pCO
2
after bicarbonate load
>20 mmHg <20 mmHg <20 mmHg <20 mmHg >20 mmHg >20 mmHg
NH
4
Cl load urine pH <5.5 >5.5 >5.5 <5.5 >5.5 <5.5
Furosemide challenge urine pH
<5.5 >5.5 >5.5 >5.5 <5.5 <5.5
Renal stones N Y N? Y Y N
Y, yes; N, no; HCMA, hyperchloremic metabolic acidosis; UAG, urinary anion gap; UOG, urinary osmolar gap; U-B pCO2, urine to bl
ood partial pressure carbon
dioxide gradient.