Geckeler K.E., Nishide H. (Eds.) Advanced Nanomaterials
Подождите немного. Документ загружается.

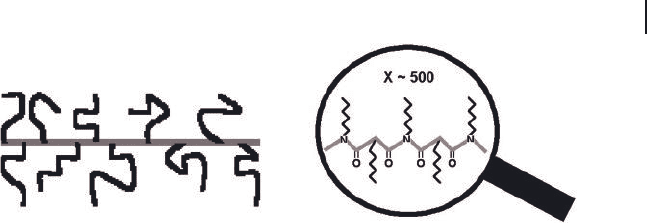
2.3 Solution Properties 75
tively. Other than a large size difference, a nanofi ber bears remarkable structural
resemblance to PHIC. Thus, block copolymer nanofi bers can be viewed as a mac-
roscopic counterpart of a polymer chain or a “ suprapolymer ” chain or “ giant ”
polymer chain [34] . In this subsection, preliminary results showing the similarities
and dissimilarities between the solution properties of polymer chains and diblock
nanofi bers will be reviewed.
To study the dilute solution properties of nanofi bers and polymer chains, the
fi bers should be made suffi ciently short, so that they remain dispersed in the
solvent for a long, or even an infi nitely long, period of time. The use of relatively
short nanofi bers also ensured their characterization by classic techniques, such as
light scattering (LS) and viscometry. While we have studied nanofi bers prepared
from several block copolymer families, for clarity, the discussion will be restricted
to PS - PI nanofi bers obtained by cross - linking cylindrical micelles of PS
130
- PI
370
formed in N , N - dimethyl acetamide [24] . The preparation of such fi bers has been
discussed previously and the right - hand panel of Figure 2.5 shows a TEM image
of the nanofi bers thus prepared in THE after aspiration onto a carbon - coated
copper grid. As the magnifi cation was known for such images, we were able to
measure manually the lengths of more than 500 fi bers for this sample. The data
from such measurements allowed us to construct the length distribution function
of this sample denoted as fraction 1 or F1 in Figure 2.11 . From the length distribu-
tion function, we obtained the weight - and number - average lengths and L
w
and
L
n
. The L
w
and L
w
/ L
n
values are 3490 nm and 1.35 for this sample.
While ultracentrifugation [34] or density gradient centrifugation could have been
used, in principle, to separate the fi bers into fractions of different lengths, we
obtained nanofi ber fractions with shorter lengths by breaking up the longer
nanofi bers by ultrasonication [26] . By adjusting the ultrasonication time, we pro-
duced fi bers of different lengths. Also shown in Figure 2.11 are the length distri-
bution functions for samples denoted as F3 and F5, which were ultrasonicated for
4 and 20 h, respectively. As ultrasonication time increased, the distribution shifted
to shorter lengths.
These fi ber fractions were suffi ciently short and allowed us to determine their
weight - average molar, M
w
, by light scattering. Figure 2.12 shows a Zimm plot
Figure 2.10 Structural comparison between a PS - PCEMA
nanofi ber (left) and a PHIC chain (right) at different
magnifi cations.

76 2 Block Copolymer Nanofi bers and Nanotubes
Figure 2.12 Zimm plot for the light scattering
data of F3 in the scattering angle range
of 12 to 30 ° . The solid circles represent the
experimental data. The hollow circles
represent the extrapolated Kc / Δ R
θ
|
c
→ 0
data.
(a) Linear extrapolation of data to zero
concentration at the highest and lowest
scattering angles of 30 and 12 ° is illustrated.
(b) The result of curve fi tting of the Kc / Δ R
θ
|
c
→ 0
data using Equation (2.1) .
Figure 2.11 Plot of fi ber population density P (L) versus length
L for PS
130
- PI
370
nanofi ber fractions 1 (
ⵧ
), 3 (
䊉
) and 5 (
䊊
)
generated from TEM image analysis.
2.3 Solution Properties 77
for the light scattering data for sample F3 in the scattering angle, θ , range of 12
to 30 ° .
The data quality appears high. Multiple runs of the same sample indicated that
the data precision was high.
For the large - sized fi bers, the Kc / Δ R
θ
data varied with sin
2
( θ /2) or the square of
the scattering wave vector q non - linearly, despite the low angles used. We fi tted
the data using Equation (2.1) :
Kc
RM
qR kqR Ac
Δ
θ
=+
()
−
[]
+
1
113 2
22 44
2
w
GG
(2.1)
and obtained M
w
, the radii of gyration R
G
and the second Virial coeffi cient A
2
for
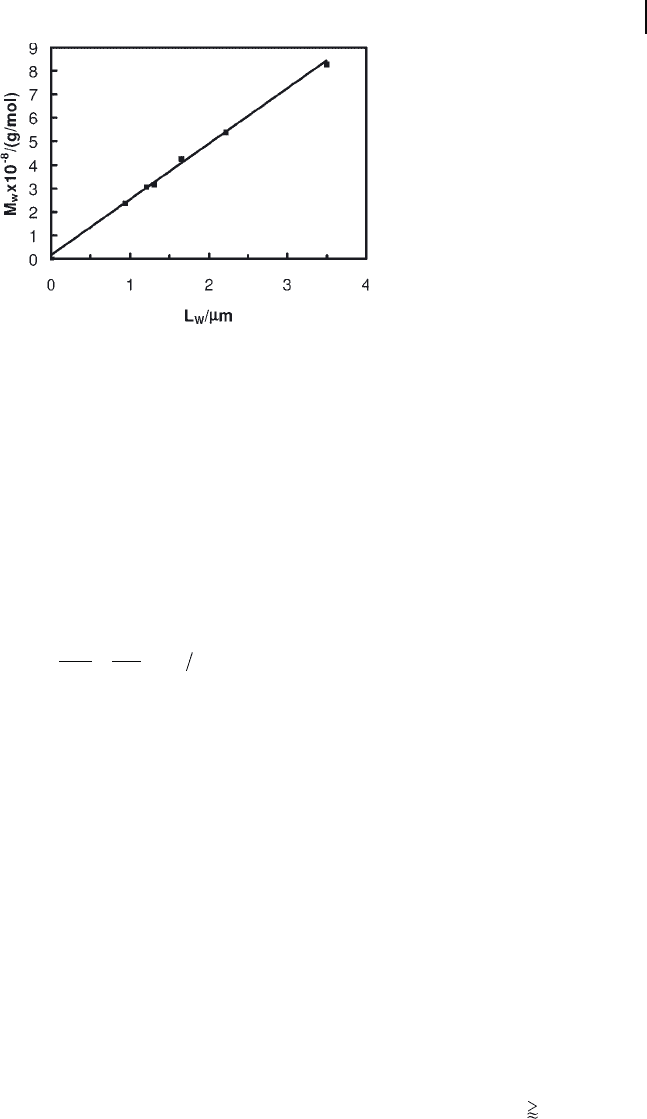
the different fractions. Figure 2.13 plots the resultant M
w
, versus L
w
, where the
values for L
w
were obtained from TEM length distribution functions P ( L ). The
linear increase in M
w
with L
w
suggests the validity of the M
w
values determined.
The validity of the M
w
value for F3 was further confi rmed recently by Professor
Chi Wu ’ s group at the Chinese University of Hong Kong, who performed a light
scattering analysis of a nanofi ber sample down to θ = 7 ° . At such low angles, the
kq R
44
G
term in Equation (2.1) was not required for curve fi tting and data analysis
by the Zirnm method should yield accurate M
w
and R
G
values.
After nanofi ber characterization, we then proceeded to check the dilute solution
viscosity properties. Our experiments indicated that the nanofi ber solutions were
analogous to polymer solutions and were shear thinning, i.e., the viscosity of a
sample decreased with increasing shear rate. This occurred for the alignment
of the nanofi bers along the shearing direction above a shear rate γ of ≈ 0.1 s
− 1
[35] . While both nanofi ber and polymer solutions are shear thinning, the fi elds
required for shear thinning are dramatically different. Polymers of ordinary molar
mass, e.g. < 10
6
g mol
−
1
, would experience shear thinning only if γ 10
− 4
s
− 1
[36] .
Figure 2.13 Increase in LS M
w
with TEM L
w
for PS
130
- PI
370
nanofi ber fractions.
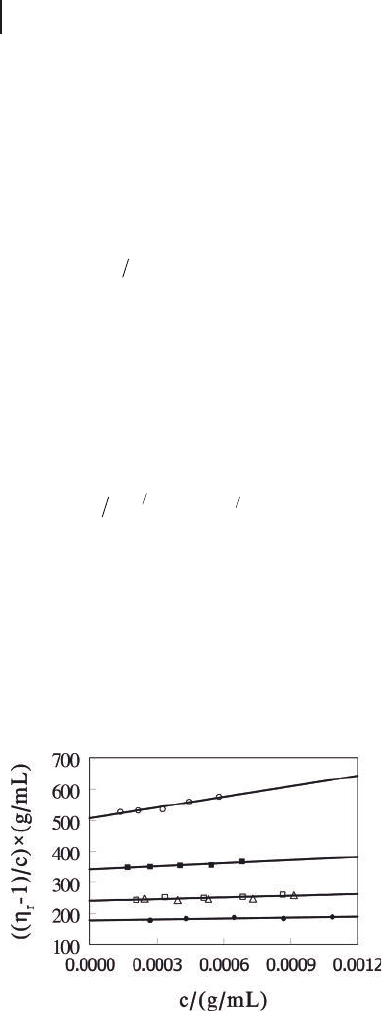
78 2 Block Copolymer Nanofi bers and Nanotubes
The huge difference should be a direct consequence of the drastically different
sizes between the two.
To minimize the shear - thinning effect, we measured the viscosities of dilute
solutions of the nanofi ber fractions in THF using a laboratory - built rotating cyl-
inder viscometer at γ = 0.082 s
−
1
[37] . Figure 2.14 shows the ( η
r
− 1)/ c data plotted
against nanofi ber concentrations c , where η
r
, the relative viscosity, is defi ned as
the ratio between the viscosities of the nanofi ber solution and solvent THF. The
solid lines represent the best fi t to the experimental data by Equation (2.2) :
ηηη
rh
−
()
=
[]
+
[]
1
2
ckc
(2.2)
where [ η ] is the intrinsic viscosity and k
h
is the Huggins coeffi cient. The linear
dependence between ( η
r
− 1)/ c and c is in striking agreement with the behavior
of polymer solutions. Even more interesting, k
h
took values mostly between 0.20
and 0.60 in agreement with those found for polymers [36] .
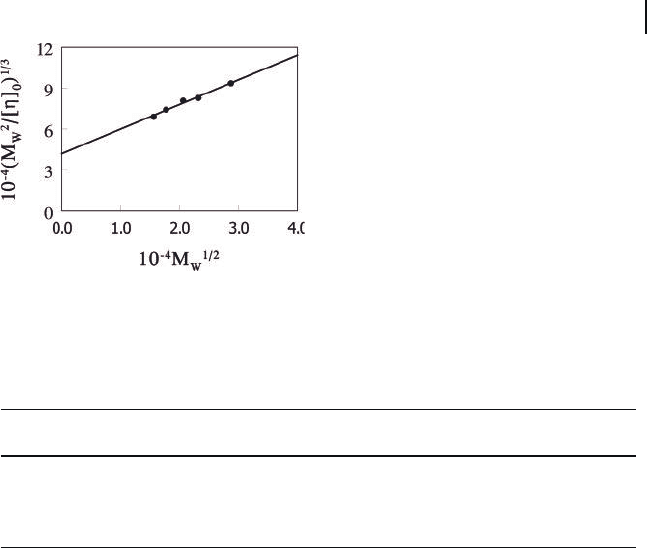
We further treated the [ η ] data with the Yamakawa – Fujii – Yoshizaki (YFY)
theory, originally developed for wormlike chains [38, 39] . According to Bohdanecky
[40] , the YFY theory could be cast in a much simpler form, Equation (2.3) :
MABM
ww
2
13
12
η
[]
()
=+
(2.3)
for chains with a wide range of reduced chain lengths. In Equation (2.3) , A and B
are fi tting parameters that are related to the persistence length l
p
and the hydro-
dynamic diameter d
h
of the chains, respectively. Figure 2.15 shows the data that
we obtained for the PS
130
- PI
370
nanofi bers in THF plotted following Equation (2.3) .
From the intercept A and slope B of the straight line, we calculated l
p
and d
h
for
the nanofi bers to be (1040 ± 150) and (69 ± 18) nm, respectively.
Figure 2.14 From top to bottom, plot of ( η
r
− 1)/ c versus c
for PS
130
- PI
370
nanofi ber fractions 2, 3, 4 and 6 in THF. All the
η
r
data were obtained using the viscometer at a shear rate of
0.082 s
− 1
with the exception of those denoted by ( Δ ), which
were obtained at a shear rate of 0.047 s
− 1
.
2.3 Solution Properties 79
This procedure was repeated for the nanofi bers in different solvents. Table 2.1
summarizes the l
p
and d
h
values that we determined in three different solvents for
the PS
130
- PI
370
nanofi bers. The d
h
value in THF compares well with what we esti-
mated from the sum between the diameter of the cross - linked PI core determined
from TEM and the root - mean - square end - to - end distance of the PS coronal chains,
and thus suggests the applicability of the FYF theory to the nanofi ber solutions.
What is more convincing is the decreasing trend for the determined d
h
values with
increasing DMF content in THF – DMF mixtures. While both THF and DMF solu-
bilize PS, d
h
decreased with increasing DMF content because the extent of swelling
for the cross - linked PI core decreased with increasing DMF content.
The l
p
values reported in Table 2.1 are comparable to those reported by Discher
and coworkers [41, 42] and by Bates and coworkers [43, 44] for PEO - PI cylindrical
micelles with a core diameter of ≈ 20 nm prepared in water, where PEO denotes
poly(ethylene oxide). While Bates and coworkers deduced the l
p
values from small -
angle neutron scattering, Discher and coworkers determined the l
p
values from
fl uorescence microscopy. In the latter case, they compared the dynamic behavior
of single cylindrical micelles before and after PI core cross - linking. After micelle
cross - linking, the micelles became much more rigid dynamically, which means
that the contour or conformation of the fi bers, in contrast to the micelles, changed
or fl exed very little with time, despite their rotation in space as approximate rigid
rotors. By performing a dynamic analysis of the fl exion motion by subtracting off
the spontaneously curved average shape of the fi bers, they concluded that the
Figure 2.15 Nanofi ber viscosity data plotted following the Bohdanecky method.
Table 2.1 Persistence length l
p
, and hydrodynamic diameter d
h
of the nanofi bers calculated from the viscosity data for
PS
130
- PI
370
nanofi ber fractions in various solvents.
Solvent d
h
/ nm l
p
/nm
THF
69 ± 18 1040 ± 150
THF − DMF = 50/50 61 ± 850 ± 90
THF − DMF = 30/70 51 ± 12 830 ± 60

80 2 Block Copolymer Nanofi bers and Nanotubes
dynamic l
p
values of the fi bers were about 50 times higher than those of the cylin-
drical micelles. From viscometry, one deduces the static l
p
values of the nanofi bers,
which measure on average how much an ensemble of fi bers bends. Therefore,
one should not compare the viscometry l
p
values determined by us with those
determined by Discher and coworkers, who totally ignored the locked - in curva-
tures of the fi bers in their analysis.
To get a clue to the static l
p
values of the PEO - PI fi bers studied by Discher and
coworkers, from their fl uorescence microscopy images we noticed that the kinks
in the original cylindrical micelles were locked in after micelle cross - linking and
the fi bers assumed conformations similar to those before micelle cross - linking.
Thus, the static l
p
values of the nanofi bers should be similar to those of the cylin-
drical micelles. The fact that the l
p
values that we determined from viscometry are
comparable to those of the PEO - PI cylindrical micelles with similar core diameters
again suggests the validity of the YFY theory in treating the nanofi ber viscosity
data.
The above study demonstrates that block copolymer nanofi bers have dilute solu-
tion properties similar to those of polymer chains. In an earlier report [45] , we also
demonstrated that block copolymer nanofi bers have concentrated solution proper-
ties similar to those of polymer chains. According to the theories of Onsager [46]
and Flory [47] , polymer chains with l
p
/ d
h
> 6 would form a liquid crystalline phase
above a critical concentration. We did show the presence of such a liquid crystal-
line phase by polarized optical microscopy for PS - PCEMA nanofi bers dissolved in
bromoform at concentrations above ≈ 25 wt - % [45] . Furthermore, we observed that
such liquid crystalline phases disappeared as the temperature was raised and the
liquid crystalline to disorder transition was fairly sharp.
While block copolymer nanofi bers behave similarly to polymer chains in many
aspects, the drastic size difference between the two dictates that they have sub-
stantial property differences. Because of the large size of the nanofi bers, they
obviously move more sluggishly. Hence, we observed that a liquid crystalline
phase was formed only after the PS - PCEMA nanofi ber solution was sheared
mechanically. Also, because of their sluggishness, the liquid crystalline phase
could not reform spontaneously after cooling a system if it had been heated above
the liquid crystalline to disorder transition temperature. Thus, we can predict,
without performing any sophisticated experiments, that the analogy between
nanofi bers and polymer chains will fail after the molar mass or the size of the
nanofi bers exceeds a critical value. As the size of the nanofi ber increases, the
gravitational force driving the settling of the nanofi ber increases and the dispers-
ibility of the nanofi ber decreases. Furthermore, the van der Waals forces between
different nanofi bers increase [48] , which can cause different nanofi bers to cluster
and settle.
We recently examined the stability of nanofi bers dispersed in THF prepared
from PS
130
- PI
370
. This particular nanofi ber sample had L
w
= 1650 nm, L
W
/ L
n
= 1.21
and M
w
= 4.3 × 1 0
8
g mol
− l
, respectively. At a concentration of ≈ 8 × 1 0
− 3
g mL
− l
and
under gentle stirring, no nanofi ber settling was observed during 4 days of observa-
tion by light scattering. Without stirring, we noticed a 10% decrease in the light

2.4 Chemical Reactions 81
scattering intensity of the solution, which corresponded to ≈ 10 wt - % settling of
the nanofi bers in the fi rst 4 days. No noticeable further settling was observed in
another 8 days [24] . This could indicate that the longer fi bers in this sample
exceeded the critical size for settling. Our light scattering and centrifugation
experiments suggested that the longer fi bers fi rst clustered and then settled. The
fact that the clustering could be prevented by gentle stirring suggests that only a
very shallow attraction potential existed between the fi bers.
Although the critical length for settling was short for this sample, several rni-
crometers, the critical length depends on many factors including the relative
length between the core and soluble block and the absolute diameter of the cores.
Methods of increasing the nanofi ber dispersity may include increasing the length
of the soluble block relative to the core block and decreasing the core diameter.
Because of the differences between the polymer chains and the nanofi bers, we
expect differences in the performances of these two classes of bulk materials.
Unfortunately, the mechanical properties of block copolymer nanofi bers or
nanofi ber composites have not been studied so far. We have not performed any
detailed studies of solution properties of block copolymer nanotubes. As a result
of the structural similarities between the two, we expect the nanofi bers and nano-
tubes to have many similar solution properties.
2.4
Chemical Reactions
The similarities between the structure and the properties of the solutions between
nanofi bers and polymer chains prompted us to ask the question as to whether
nanofi bers and nanotubes would have chemical reaction patterns similar to those
of polymer chains. A PI chain can be hydrogenated via “ backbone modifi cation ”
to yield a polyolefi n chain. Through techniques such as anionic polymerization,
etc., one can readily prepare “ end - functionalized ” polymers. The end - groups can
be further derivatized or used for additional end - fi inctionalization. This section
will show that block copolymer nanotubes can also undergo backbone modifi ca-
tion and end - functionalization.
2.4.1
Backbone Modifi cation
Backbone modifi cation has already been involved to convert triblock nanofi bers
into nanotubes. Apart from the performance of organic reactions to the nanofi bers
and nanotubes, this sub - section discusses the performance of inorganic reactions
in the cores of the nanotubes to convert them into polymer – inorganic hybrid
nanofi bers. Block copolymer nanofi bers and nanotubes are soft materials. They
will most probably fi nd applications in bio - related disciplines, such as in the
medical, pharmaceutical and cosmetic industries. For applications in nanoelec-
tronic devices, polymer – inorganic hybrid nanofi bers would be more desirable [49,

82 2 Block Copolymer Nanofi bers and Nanotubes
50] . The fi rst report on the preparation of block copolymer – inorganic hybrid
nanofi bers appeared in 2001, which dealt with fi lling of the core of the PS - PCEMA -
PAA nanotubes by γ - Fe
2
O
3
[10] .
The preparation fi rst involved the equilibration between the nanotubes and
FeCl
2
in THF. Fe(II) entered the nanotube core to bind with the core carboxyl
groups. The extraneous FeCl
2
was then removed by precipitating the Fe(II) -
containing nanotubes into methanol. Adding NaOH dissolved in THF containing
2 vol - % of water precipitated Fe(II) trapped in the nanotube core as ferrous oxide.
The ferrous oxide was subsequently oxidized to γ - Fe
2
O
3
via the addition of hydro-
gen peroxide [51] . The top panel in Figure 2.16 shows a TEM image of the hybrid
nanofi bers. The γ - Fe
2
O
3
particles can be seen to be produced exclusively inside the
nanotube cores.
The production of γ - Fe
2
O
3
in the confi ned space of the “ nanotest - tubes ” resulted
in particles that were nanometer - sized. Hence the particles were superparamag-
Figure 2.16 Top: TEM image of PS - PCEMA - PAA/Fe
2
O
3
hybrid
nanofi bers. Bottom: Bundling and alignment of the nanofi bers
in a magnetic fi eld. The arrow indicates the magnetic fi eld
direction.

2.4 Chemical Reactions 83
netic, as demonstrated by the results of our magnetic property measurement [10] .
This meant that they were magnetized only in the presence of an external magnetic
fi eld and were demagnetized when the fi eld was removed. To see how such fi bers
behaved in a solvent in a magnetic fi eld, we dispersed the fi bers in a solvent
mixture consisting of THF, styrene, divinylbenzene, and a free radical initiator
AIBN. The fi ber dispersion was then dispensed into an NMR tube and mounted
in the sample holder of an NMR instrument. In the 4.7 - T magnetic fi eld of the
NMR, the solvent phase was gelled by raising the temperature to 70 ° C to polymer-
ize styrene and divinylbenzene. Thin sections were obtained from the gelled
sample by ultramicrotoming. Shown in the bottom panel of Figure 2.16 is a TEM
image of nanofi bers in a gelled sample. One consequence of the induced magneti-
zation of the fi bers is that they attracted one another and bundled in a magnetic
fi eld. Also clear from this image is that the fi bers aligned along the magnetic fi eld
direction.
The bundling and alignment of the hybrid nanofi bers in a magnetic fi eld have
important practical implications. For example, the controlled bundling of several
nanofi bers may form the basis of magnetic nanomechnical devices. For the
construction of water - dispersible magnetic nanomechanical devices, the super-
paramagnetic nanofi bers need to be water dispersible. We recently prepared water -
dispersible polymer – Pd hybrid catalytic nanofi bers from a tetrablock copolymer
[52] and more recently polymer – Pd – Ni superparamagnetic nanofi bers from a
triblocic copolymer [53] . The tetrablock that we used was PI - P t BA - P(CEMA -
HEMA) - PGMA, where PGMA, being water soluble, denotes poly(glyceryl
methacrylate) and P(CEMA - HEMA) denotes a random copolymer of CEMA and
2 - hydroxyethyl methacrylate. The hydroxyl groups of the precursory PHEMA block
was not fully cinnamated because P(CEMA - HEMA) facilitated the transportation
of Pd
2+
and Ni
2+
.
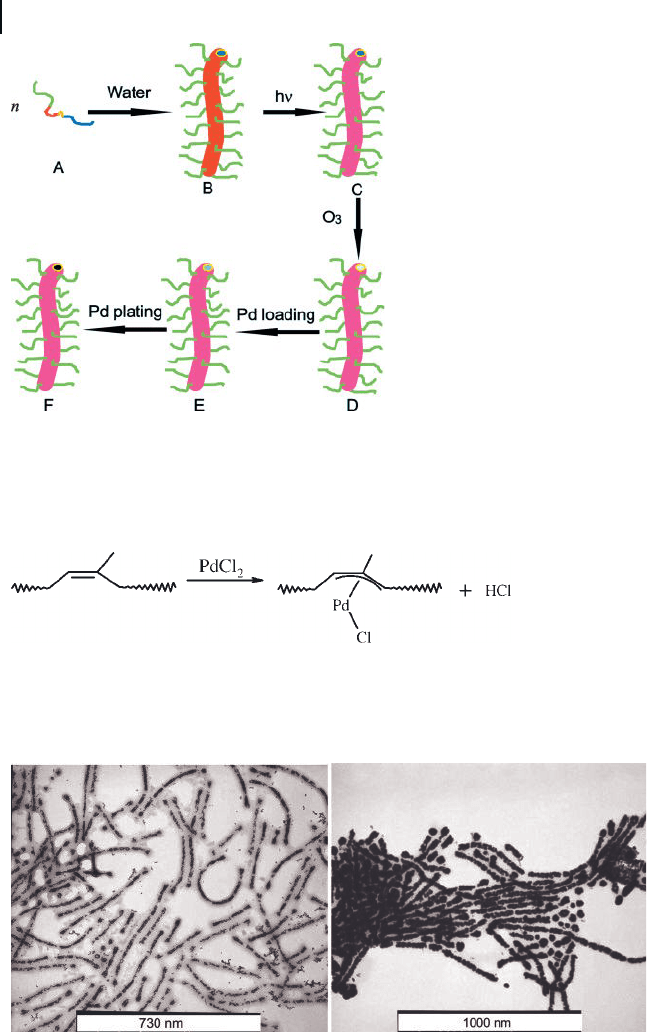
We prepared the polymer – Pd hybrid nanofi bers following the scheme depicted
in Figure 2.17 [52] . This involved fi rst dispersing freshly - prepared PI - P t BA -
P(CEMA - HEMA) - PGMA in water to yield cylindrical aggregates (A → B in Figure
2.17 ). Such aggregates consisted of a PGMA corona and a PI core. Sandwiched
between these two layers are a thin P t BA layer and a P(CEMA - HEMA) layer. Such
cylindrical aggregates were then irradiated to cross - link the (CEMA - HEMA) layer
(B → C). The PI core was degraded by ozonolysis (C → D). By controlling the
ozonolysis time, we could control the degree of PI degradation. When not fully
degraded, the residual double bonds of the PI fragments trapped inside the nano-
tubular core were able to sorb Pd(II), most probably via π - allyl complex formation,
Scheme 2.3 .
The complexed Pd(II) was then reduced by NaBH
4
to Pd (D → E). The left panel
of Figure 2.18 is a TEM image for such nanotubes containing 4.0 wt - % reduced
Pd nanoparticles. The Pd - loaded nanofi bers were dispersible in water where
many water - based electroless plating reactions occur. Thus, Pd could serve as a
catalyst for the further electroless deposition of other metals. We, for example,
loaded more Pd into the tubular core via electroless Pd plating onto the initially
formed Pd nanoparticles to yield essentially continuous Pd nanowires (E → F ,
84 2 Block Copolymer Nanofi bers and Nanotubes
Figure 2.18 TEM image of nanofi bers containing 4 wt - % Pd
(left); TEM image of nanofi bers containing 18.4 wt - % Pd
(right). The scale bars in the form of white boxes are 730 and
1000 nm long, respectively.
Figure 2.17 Schematic illustration of the processes involved
to produce water - dispersible polymer – Pd hybrid nanofi bers.
Scheme 2.3