Faulon J.L., Bender A. Handbook of Chemoinformatics Algorithms
Подождите немного. Документ загружается.

318 Handbook of Chemoinformatics Algorithms
11.2 THE CHALLENGES OF GENERATING NETWORKS
Provided an ensemble of possible chemical reactions, the network generation pro-
cess essentially consists of finding all species that can be synthesized from an initial
set of reactants. The process is recursive as reactions can be applied to generated
species as well. The process ends depending on the goal of the study, for instance,
a specific compound has been produced, or all compounds with predefined physi-
cal, chemical, or biological properties have been generated. The same process can
be used to search for all species producing a given target when reversing the reac-
tions. This reverse process is used in retrosynthesis analysis where reversed reactions
are named transforms, and species generated are named synthons [1]. The network
generation algorithms given in this chapter apply to both synthesis and retrosynthe-
sis as it is just a matter of defining the direction of the reactions. As reviewed by
Ugi et al. [2], three main approaches have been developed for network generation:
empirical approaches, whose strategies are based on data from reaction libraries,
semiformal approaches based on heuristic algorithms, where reactions are derived
from a few mechanistic elementary reactions, and formal techniques, based on graph
theory. This chapter focuses on the last approach. The formal approach has spurred
most of the network generation algorithms, which historically started with the work
of Corey et al. [3] and the retrosynthesis code Lhasa (http://www.lhasalimited.org).
Ugi et al. argue that formal techniques are general enough to be applicable to
any type of reacting system in organic or inorganic chemistry; furthermore, formal
techniques are the only methods capable of generating new reaction mechanisms
and therefore elucidating unresolved chemical processes such as those found with
synthesis planning.
While formal techniques are robust, their computational scaling limits their appli-
cability to real reacting systems. Indeed, as has been shown by several authors [4–9],
for many processes related to retrosynthesis, combustion, and petroleum refining, the
number of reactions and intermediate species that are created generally scales expo-
nentially with the number of atoms of the reactants. With metabolic and signaling
networks, the number of possible species also grows prohibitively [10,11] when one
considers all the possible states a species can fall into (phosphorylation at several
sites, and complex formation with other proteins and ligands). As a matter of fact
it has been shown that even simple models of the epidermal growth factor (EGF)
receptor signaling network can generate more than 10
23
different species [12].
With all these systems, not only the time taken to generate networks may scale
exponentially, but simulating the dynamics of large networks may also become com-
putationally prohibitive. The most common approach to study the dynamics of a
reaction network is to calculate species concentration over time by integrating a sys-
tem of ordinary differential equations (ODEs). The computational cost of integrating
ODEs depends nonlinearly on N, the number of chemical species. For stiff ODEs
(cf. definition in Section 11.4), that cost scales N
3
, and thus simulating a system for
which N is larger than 10
4
−10
5
becomes impractical.
An alternative to palliate the exponential scaling problem of formal techniques
is the reduction of the reaction mechanism. The question raised when the reduc-
ing mechanism is used is how to choose a reaction subset that describes correctly
Reaction Network Generation 319
the dynamics of the entire system. Reduction strategies in the area of combustion
modeling have been reviewed by Frenklach [6]; these are quite general and applicable
to other fields. According to Frenklach, there are five types of reduction strategies:
(1) global reduction, (2) response modeling, (3) chemical lumping, (4) statistical
lumping, and (5) detailed reduction. Global modeling techniques transform a com-
plete reaction scheme into a small number of global reaction steps. Global techniques
comprise ad hoc methods such as empirical fitting, reduction by approximations,
and lumping. All global techniques are specific to a particular problem and cannot
be generalized. Response modeling techniques consist of mapping model responses
and model variables through functional relationships. Generally, model responses are
species concentrations, and model variables are the initial boundary conditions of the
reacting mixture and the model parameters, such as rate coefficients, and transport
properties. Usually, model responses are expressedas simple algebraic functions (such
as polynomials) in terms of model variables. As with global techniques, response
modeling solutions are problem specific since they require data to build algebraic
functions. Chemical lumping was first developed for polymerization-type reactions.
Chemical lumping models are used when a polymer grows by reaction between the
polymer and monomer species. The lumping strategy is guided by similarity in chem-
ical structure or chemical reactivity of the reacting species. The main assumption of
chemical lumping is that the reactions describing the polymer growth are essen-
tially the same and the associated thermochemical and rate parameters exhibit only
a weak dependence on the polymer size. Finally, the detailed reduction technique
consists of identifying and removing noncontributing reactions. An effective reduc-
tion strategy is to compare the individual reaction rates with the rate of a chosen
reference reaction. The reference reaction is, for instance, the rate-limiting step or the
fastest reaction. The detailed reaction reduction approach is a general technique and is
a priori applicable to any reacting system.
The goal of this chapter is to present reaction network generation techniques
that are computationally tractable and general enough to be applicable to many
processes, such as synthesis planning, combustion, petroleum refining, and sig-
naling and metabolic network inferences. Thus, network generation algorithms
are presented focusing on the formal generation approach since it is the only
approach that is general enough and is applicable to different processes. As men-
tioned above, formal techniques scale exponentially with the problem size and
therefore reduction methods need to be applied. The only reduction technique appli-
cable to general systems is the detailed reduction method. Because the networks
generated can be exponentially large, network generation and reduction cannot
be performed in sequence but simultaneously. Section 11.6 presents algorithms
where the detailed reduction technique is applied “on the fly” while generating
networks.
11.3 REPRESENTATION OF CHEMICAL REACTIONS
There are several reasons why one wants to codify chemical reactions. Reactions
need to be stored and retrieved from databases and these databases often require us
320 Handbook of Chemoinformatics Algorithms
to link information from different sources. There is also a need for automatic proce-
dures for analyzing, performing correlations, and classifying reactions in databases,
a prominent example being the classification of enzymatic reactions [13]. Finally,
reactions need to be coded for the construction of knowledge bases for reaction pre-
dictions with the purpose of network generation or synthesis design. The reaction
codifications systems presented next are focusing on this latter application.
11.3.1 REPRESENTATION BASED ON REACTION CENTERS
The traditional method to represent a chemical reaction is based on reaction cen-
ters. A reaction center is constituted of the atoms and bonds directly involved in the
bond and electron rearrangement process. The reaction centers are thus composed
of the bonds broken and formed when the reaction takes place as well as the atoms
attached to these bonds. It is easy to detect these reaction centers from the reaction
graphs presented in Chapter 1, and an automated procedure to perform this task has
been worked out for some time. Among the published solutions are the ClassCode
Infochem system (http://infochem.de/en/products/software/classify.html), HORACE
[14], and the reaction classification numbers used in the KEGG database [13]. In
the solution adopted with KEGG, reaction centers are searched between all possible
pairs (reactant × product) involved in a reaction. An example of detection of reaction
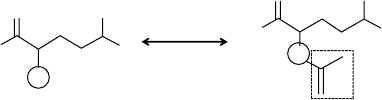
center is depicted in Figure 11.1.
To find the reaction center between the elements of a given pair, one first searches
the maximum common substructure between the elements. Next, each element (reac-
tant and product) is decomposed into a common part and a nonoverlapping part
(dashed boxes in Figure 11.1). The reaction center is the atom (circled in Figure 11.1)
belonging to the common part and adjacent to the nonoverlapping part. The process
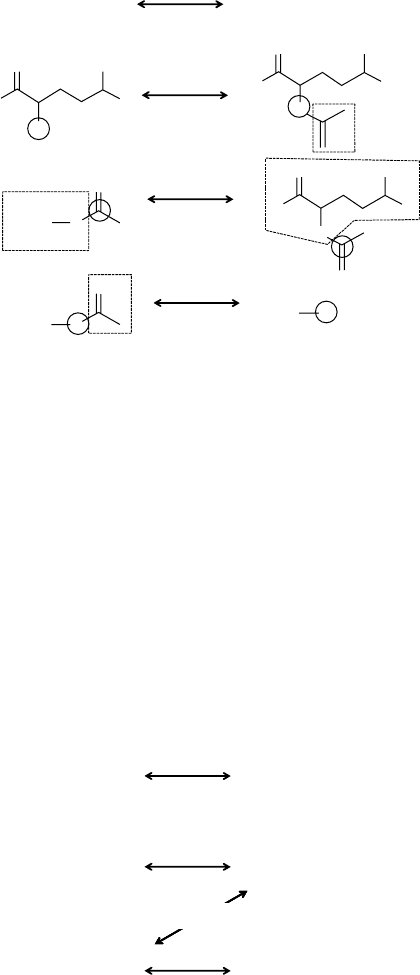
is repeated between all possible pairs and as illustrated in Figure 11.2, a given reac-
tion is characterized by several reaction centers, common parts, and nonoverlapping
parts. All reaction centers (R), nonoverlapping (D) parts, and common parts (M) of
the KEGG database have been numbered uniquely; thus each reactant ×product pair
is characterized by three numbers R, D and M, also named the RC numbers. For
instance, for the glutamate ×N-acetyl-glutamate, R = 173, D = 349 and M = 14;
the RC number for the pair is thus 173.349.14. As illustrated in Figure 11.3, when
several reactant ×product are found for a reaction, the code for the reaction is the
compilation of the RC numbers of all pairs.
173.349.14
Glutamate
N-acetyl-
glutamate
OO
N
O
O
O
O
O
N
O
FIGURE 11.1 Reaction center between glutamate and N-acetyl-glutamine. The reaction cen-
ters are circles, the nonoverlapping part is in a dashed box, and the remaining parts of the
structures are the common parts. The reaction center (circled) is the atom in the common part
adjacent to the nonoverlapping part.
Reaction Network Generation 321
61.718.8
298.349.8
Glutamate + acetyl-CoA N-acetyl-glutamate + CoA
N-acetyl-
glutamate
Acetyl-CoA
Acetyl-CoA
O
O O
OO
N
CoA S
CoA
CoA
S
CoA S
173.349.14
Glutamate
N-acetyl-
glutamate
OO
N
O
O
O
O
O
N
O
FIGURE 11.2 Reaction classification for amino acid N-acetyltransferase (EC 2.3.1.1)
between all pair reactants (glutamate, acetyl-Co Enzyme A) × products (N-acetyl-glutamate,
Co Enzyme A). The figure depicts reaction centers, nonoverlapping parts, and common parts.
All parts are depicted using the notation of Figure 11.1.
11.3.2 BOND–ELECTRON MATRICES
Bond–electron matrices were first introduced by Ugi and coworkers [15,16]. In
this representation, compounds and reactions are coded by bond–electron (be-) and
reaction (r-) matrices (cf. Figure 11.4). In a given be-matrix representing a com-
pound, the ith row (and column) is assigned to the ith atom of the compound. The
entry b
ij
,(i = j)ofabe-matrix is the bond order of the covalent bond between atoms
i and j. The diagonal entry b
ii
is the number of free valence electrons for atom i.Itis
worth noting that the adjacency and connectivity matrices (cf. Chapter 1) differ from
the be-matrices in their diagonal entries. The redistribution of valence electrons by
173.349.14
298.349.8
Glutamate + acetyl-CoA
1. Glutamate
2. Acetyl-CoA
N-acetyl-glutamate + CoA
3. N-acetyl-glutamate
4. CoA
61.718.8
FIGURE 11.3 Codification classification for amino acid N-acetyltransferase (EC 2.3.1.1).
The code reflects all mapping between reactant and products. The mappings are detailed in
Figure 11.2, and the resulting code is 1_3(173.349.14) +2_3(61.718.8) +2_4(298.349.8).
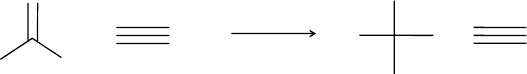
322 Handbook of Chemoinformatics Algorithms
2
34
4
5
–
–
656
+
OO
HH
NH
2
1
C
NC
H
1
3
B matrix
C
1
C
1
021100
200000
100000
100000
000013
000030
O
2
O
2
H
3
H
3
H
4
H
4
C
5
C
5
N
6
N
6
R matrix
C
1
C
1
0–1 0 0 1 0
–110000
000000
000000
1000–10
000000
O
2
O
2
H
3
H
3
H
4
H
4
C
5
C
5
N
6
N
6
E matrix
C
1
C
1
011110
110000
100000
100000
100000
000030
O
2
O
2
H
3
H
3
H
4
H
4
C
5
C
5
N
6
N
6
FIGURE 11.4 Example of bond–electron (be-) matrices and reaction (r−) matrices. Negative
numbers in the reaction matrix represent broken bonds while positive numbers represent bonds
that are created as the reaction proceeds. The sum of a row (column) of be-matrices equals the
valence of the corresponding atoms. The sum of a row (column) of r-matrices equals zero.
which the starting materials of chemical reactions are converted into their products is
represented by r-matrices.
The fundamental equation of the Dugundji–Ugi model for any given reaction α
1
+
α
2
+···+α
n
→ β
1
+β
2
+···+β
n
is B +R = E, where B (Begin) and E (End) are
the be-matrices of reactants and products and R is the reaction matrix. The addition
of matrices proceeds entry by entry, that is, b
ij
+r
ij
= e
ij
. Since there are no formal
negative bond orders or negative numbers of valence electrons, the negative entries
of R must be matched by positive entries of B of at least equal values.
11.3.3 REPRESENTATION BASED ON FINGERPRINTS
Several reaction codification systems are based on molecular fingerprints: examples
include the Daylight reaction fingerprints (http://www.daylight.com/dayhtml/doc/
theory/theory.finger.html), the reaction signature [5,17], and fingerprints based on
atom types [18]. These codification systems have in common that the fingerprint of
the reaction is the difference between the fingerprints of the products and the reac-
tants. Taking the example of signature, let us recall from Chapters 3 and 9 that the
signature of a molecule (reactant or product) is the sum of the signatures of all atoms
of that molecule. The signature of an atom x,
h
σ
G
(x), in a molecule G is a subgraph
containing the neighborhood of that atom up to a certain distance, named signature
height. Examples of atom and molecular signatures are given in Figure 11.5.
Let B and E be two molecular graphs representing the reactants and products of
the reaction R : B → E. Note that signatures can be computed on graphs that are not
necessarily connected; hence B and E can both be composed of several molecules,
including severalcopies of the samemolecule to reflect stoichiometry. The h-signature
Reaction Network Generation 323
HO
CH
2
(a) Atom signature
(b) Molecular signature
[C] ( [C ] = [C] [O] )
[C] ( [C] ( = [C] ) = [C] ( [C] )
[C] ( [C] ( = [C] ( [C] ) ) = [C] ( [C] ( = [C] ) ) [O] ( [H] )
h=1
σ (Tyrosine) =
[C] ( [C] = [C] [O] ) + 4 [C] ( [C] = [C] ) + [C] ( [C] [C] = [C] ) + [O] ( [C] [H] ) + [C] ( [C] [H] [H] )
h=1
[C]h=0
h=2
h=3
C
C
C
C
C
C
[O] ( [H] ) )
FIGURE 11.5 Atom and molecular signatures. (a) The signature is given for the red carbon
atom. (b) The molecular signature of tyrosine is derived by summing the signatures of all atoms
(in the example, signatures are not computed for hydrogen atoms).
of reaction R is given by the equation
h
σ(R) =
h
σ(E) −
h
σ(B). (11.1)
An example of reaction signature is given in Figure 11.6.
3.1.1.1
1.0 [R] ( [O] ) 1.0 [O] ( [H] [H] )
1.0 [R] ( [C] )
1.0 [R] ( [O] )
1.0 [O] ([R] [H] )
1.0 [O] ( [=C] )
1.0 [O] ( [R] [C] )
1.0 [R] ( [C] )
1.0 [O] ( [=C] )
1.0 [O–] ( [C] )
1.0 [C] ( [=O] [O– ] [R ] )
1.0
σ(3.1.1.1)=σ(Alcohol)+σ(Carboxylate)-σ(Carboxylic ester)-σ(H
2
O)
[C] ( [=O] [O] [R] )
–1.0 [O] ([ [R] [C] )
–1.0 [O] ( [H] [H] )
1.0 [O–] ( [C] )
1.0 [O] ( [R] [H] )
–1.0 [C] ( [=O] [O] [R] )
1.0 [C] ( [=O] [O–] [R] )
Ο
RO
O
RO
–
R
++
OH
2
ROH
FIGURE 11.6 Signature for the carboxylesterase enzymatic reaction (EC number 3.1.1.1).

324 Handbook of Chemoinformatics Algorithms
11.4 NETWORK KINETICS
In order to study the kinetics of networks produced by a reaction network generator
to, for instance, calculate species concentrations versus time, one needs to estimate
reaction rates for each reaction and then use these rates to simulate the kinetics.
Methods that have been proposed to perform these tasks are presented next. The
presentation is limited to techniques that have been coupled with network generation;
these methods are thus constrained by the fact that generated networks may be large;
in other words, the techniques must be computational efficient.
11.4.1 ESTIMATING REACTION RATES
In chemical kinetics the rate of a reaction allows one to compute the amount of reactant
lost and product formed during an infinitesimal small time interval dt. Considering the
chemical reaction aA +bB → cC +dD, where a, b, c, and d are the stochiometric
coefficients, the concentrations [A], [B], [C], and [D] of the reactants and products
are derived from the following equation:
−
1
a
d[A]
dt
=−
1
b
d[B]
dt
=
1
c
d[C]
dt
=
1
d
d[D]
dt
= r, (11.2)
where r is the reaction rate. The reaction rate is in turn calculated with the expression:
r = k(T)[A]
n
[B]
m
, (11.3)
where k(T) is the rate constant depending on temperature T, and n and m are
reaction orders linked to the reaction mechanism. For instance, for first-order reac-
tion of the type A → C +D, n = 1 and m = 0; for second-order reaction of the
type A +A → C +D, n = 2 and m = 0; and for second-order reaction of the type
A +B → C +D, n = 1 and m = 1. After integrating Equation 11.2, one is able to
compute the concentrations of the species versus time from the initial concentrations.
Assuming that the initial concentrations and the reaction orders are known, the only
parameter that remains to be computed is the rate constant. While some rate con-
stants along with other kinetics parameters can be found in databases [for instance,
NIST thermodynamics database for chemicals (http://webbook.nist.gov), BRENDA
(http://www.brenda-enzymes.org/) for enzyme catalyzed reactions], only a limited
number of reactions are stored in these databases, and there is a need to automatically
compute rate constants, especially when generating networks. To this end, several
approaches have been developed. One approach makes use of an approximation based
on the Arrhenius equation:
k(T) = A exp
−
E
a
RT
, (11.4)
where R is the gas constant, A is the so-called pre-exponential factor, and E
a
is the
activation energy. In this approach, which has been utilized for thermal cracking [5,7],
the pre-exponential factor is compiled from experimental literature data for five reac-
tion classes (also named elementary transformations) independent of species-specific

Reaction Network Generation 325
Homolysis:
R1 0 1
R2 1 0
R
1
-R
2
-> R
1
• + R
2
•
B-matrix
1–1
–1 1
R-matrix
01
01
Recombination:
R1 1 0
R2 0 1
R
1
• + R
2
• -> R
1
-R
2
–1 1
1–1
01
10
H-abstraction:
H01
C10
0
0
CH + R• -> RH + C•
0–1
–1 1
1
0
00
01
1
0
R 0 0 1 1 0 –1 1 0 0
β-scission:
R00
C
α
01
1
1
R-C
β
-C
α
• -> C
α
= C
β
+ R•
10
0–1
–1
1
10
00
0
2
C
β
110–110 020
Addition:
R10
C
α
00
0
2
C
α
= C
β
+ R• -> R-C
β
-C
α
•
–1 0
01
1
–1
00
01
1
1
C
β
020 1–10 110
E-matrix
FIGURE 11.7 The five elementary transformations of hydrocarbon thermal cracking used in
Ref. [5] and their associated (be-) and (r-) matrices.
structures. The elementary transformations are bond homolysis, radical recombina-
tion, hydrogen abstraction, b-scission, and b-addition (cf. Section 11.5 and Figure 11.7
for further explanations). In the approach proposed by Susnow et al. [7], the activa-
tion energy is species specific and is computed from linear free energy relationships
(LFERs) relating the activation energy to the heat of the reaction. The heat of reactions
is either retrieved from the NIST thermodynamics database or calculated using the
semiempirical molecular orbital package (MOPAC) (http://openmopac.net/).
With the goal of producing more accurate rate constants another approach has
been developed based on a group additivity method. In that method the rate constant
is related with compound thermodynamics properties (heat of formation and entropy)
by the following equation:
k(T) = C exp
−
ΔH
RT
exp
ΔS
R
, (11.5)
where C is a constant depending on temperature, molar volume, and reaction path, and
ΔH and ΔS are, respectively, the enthalpy and entropy differences between the tran-
sition states geometries and the reactants states. Entropy and enthalpy are calculated
using group additivity. The main idea of computing thermodynamics properties by
summing the properties of individual groups was initially developed by Benson and
Buss, as described in [19]. In the Benson and Buss method, the atom properties are
compiled for every possible configuration an atom can have with its first neighbors,
and the property of the molecule is simply the sum of the properties of each atom. For
instance, using the signature notation of Figure 11.5, alkanes (including methane) have
only five possible groups [C]([H][H][H][H]), [C]([C][H][H][H]), [C]([C][C][H][H]),
[C]([C][C][C][H]), and [C]([C][C][C][C]), and the property of every alkane is simply
326 Handbook of Chemoinformatics Algorithms
the scalar product between the molecular signature of the compound and the calcu-
lated property of the groups. To compute group properties, quantum calculation can
be used. As an example of application of the group additivity method relevant to the
chapter, Sumathi et al. [20,21] calculated enthalpies, entropies, and heat capacities
for hydrocarbon–hydrogen abstractions using ab initio calculations.
11.4.2 SIMULATING NETWORK KINETICS
Once rates have been estimated, the species concentrations can be calculated inte-
grating reaction equations such as Equation 11.2. There are essentially two main
mathematical approaches to integrate a system of reaction equations. In the first
approach, the concentrations of chemical species are modeled as continuous vari-
ables that change over time and reactions between species are modeled by ODEs.
The set of ODEs are often numerically integrated using solvers that can be found in
many freeware and commercial packages. In the second approach, chemical species
are treated as discrete variables that change over time in a discrete manner. In this
approach, reactions are individual events that update the system and can be combined
into a chemical master equation [22]. The chemical master equation is often compu-
tationally integrated to compute the time evolution of the systems using stochastic
simulation algorithm (SSA). Both method (ODEs and SSA) provide exact solutions
albeit there are instances where ODEs break down. An example of such a breakdown
with ODEs is when an individual reaction event causes a large difference in the likeli-
hood that other reactions will occur, and so the precise order and timing of individual
reactions can influence the overall system behavior. ODEs have also difficulties to
solve stiff equations, that is, a differential equation for which numerical methods are
numerically unstable, unless the step size is taken to be extremely small. For the above
reasons, all algorithms presented in the chapter make use of SSAs rather than ODEs.
SSAs were first proposed by Gillespie [22] and are based on Monte Carlo (MC)
sampling. The MC–Gillespie technique monitors the number of particles for each
species versus time. The initial number of particles of the reactants is computed
from their initial concentrations (given by the user) and the initial number, M
p
,of
particles in the system. In the present chapter, the MC–Gillespie technique is used at
constant volume V, which is calculated from the initial number of particles and the
particle density (both user inputs). The MC–Gillespie technique is an exact method
for numerical integration of the time evolution of any spatially homogeneous mixture
of molecular species that interact through a specified set of coupled chemical reaction
channels. The technique is based on a fundamental equation giving the probability
at time t that the next reaction in V will occur in the differential time interval [t +τ,
t +τ +dτ] and will be an r
μ
reaction:
P(τ, μ)dτ = P
1
(τ)P
2
(μ|τ), (11.6)
where P
1
is the probability that the next reaction will occur between times t +τ and
t +τ +dτ:
P
1
(τ) = a exp(−aτ)dτ, (11.7)

Reaction Network Generation 327
and P
2
is the probability that the next reaction will be r
μ
:
P
2
(μ|τ) =
a
μ
a
(11.8)
with
a = Σ
μ
a
μ
. (11.9)
In the previous equations, a
μ
dτ is the probability, to first order in dτ, that a reaction
r
μ
will occur in V in the next time interval dτ. The rate constant k
μ
is related to a
μ
in
different ways depending on whether the reaction is monomolecular or bimolecular.
For monomolecular reactions
a
μ
=[s]k
μ
, (11.10)
where [s] is the number of particles of reactant species s. For bimolecular reactions
involving two species s1 and s2,
a
μ
=[s
1
][s
2
]
k
μ
V
, (11.11)
and for bimolecular reactions involving only one reactant species,
a
μ
=[s]([s]−1)
k
μ
2V
. (11.12)
In order to integrate Equation 11.6, Gillespie proposes the following scheme. First,
generate a random value t according to P
1
(t) and, second, generate a random integer
μ according to P
2
(μ|τ). The random value τ is computed by simply drawing a random
number r
1
from a uniform distribution in the unit interval and taking
τ =
1
a
ln
1
r
1
. (11.13)
In turn, the random integer μ is generated by drawing another random number r
2
in the unit interval and by taking μ the smallest integer verifying
μ
ν=1
a
ν
> r
2
a. (11.14)
In his original paper, Gillespie [22] has proven that expressions 11.13 and 11.14
are indeed correct to simulate stochastically the time evolution of homogeneous
reactions. Since this original paper, improvement in SSAs has appeared in the lit-
erature, especially regarding computational time; these have recently been reviewed
by Gillespie [23].