Албертс Б., Брей Д. и др. Молекулярная биология клетки. Том 1
Подождите немного. Документ загружается.

141
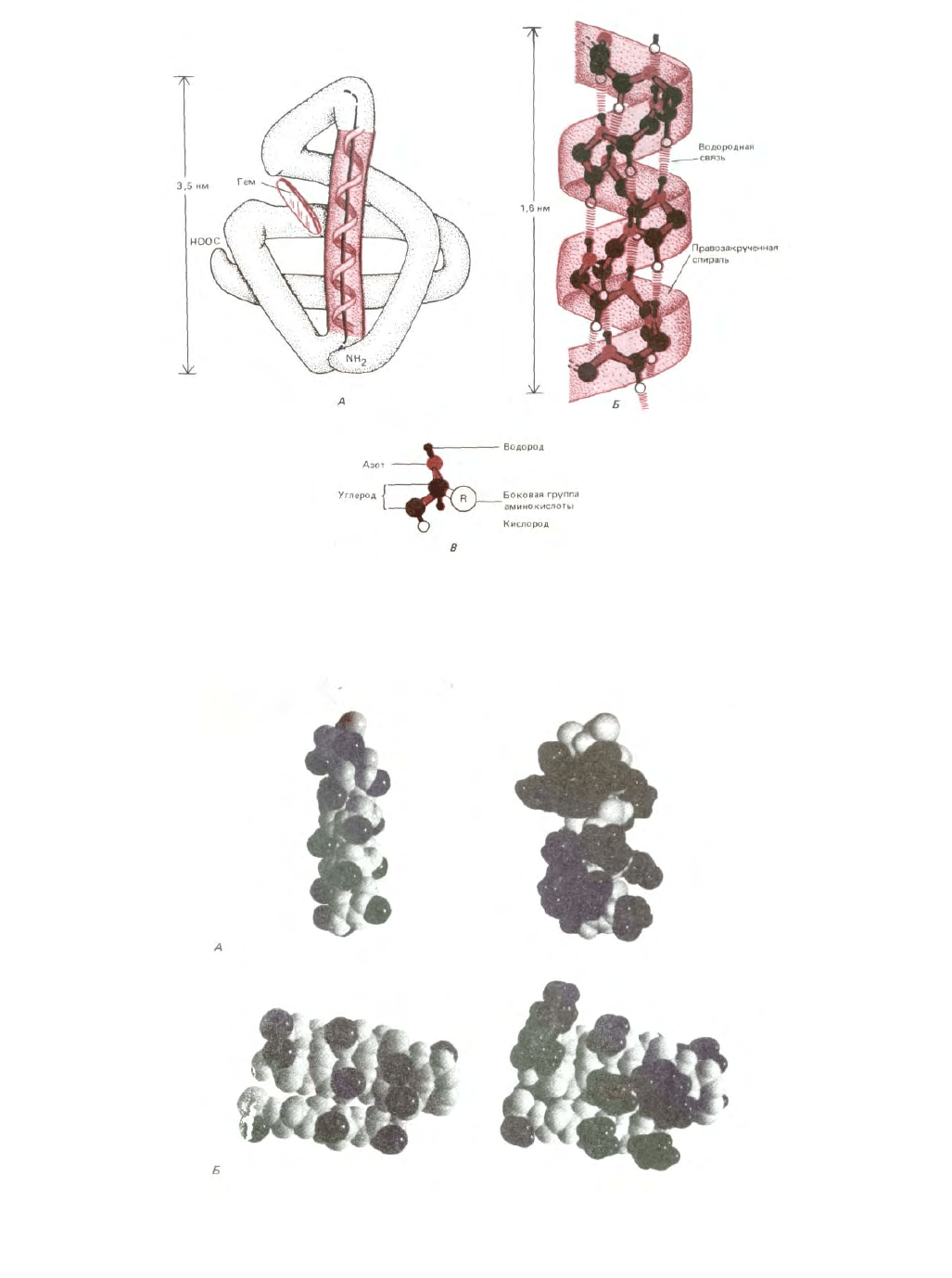
Рис. 3-26. α-Спираль - еще одна общая структура, обычно образующаяся в отдельных участках полипептидной цепи белков. А. Показана
переносящая кислород молекула миоглобина (длиной 153 аминокислоты); один из α-спиральиых участков выделен цветом. Б. Детальное
изображение совершенной α-спирали. Как и в β-слое, каждая пептидная группа связана с соседними пептидными группами водородными связями.
В. Атомы в аминокислотном остатке. Заметим, что в Б боковые группы аминокислот для упрощения опущены (они расположены на наружной
поверхности спирали. В В они обозначены как R на атоме α-углерода каждой аминокислоты (см. также рис. 3-27).
Рис. 3-27. Пространственные модели α-спирали и β-СЛОЯ. Слева структуры показаны без боковых групп аминокислот, справа - с боковыми
группами. А. α-Спираль (часть структуры миоглобина). Б. Участок β-СЛОЯ (часть структуры домена иммуноглобулина). На фотографиях слева
каждая поверхность цепи представлена только одним черным атомом (группы R на рис. 3-25 и рис. 3-26); вся поверхность цепи показана справа. (С
любезного разрешения Richard J. Feldmann.)
142
клеточных волокнах α-кератина, обусловливающих прочность кожи. Пространственные модели α-спирали и β-складчатого слоя белков с боковыми
группами и без них представлены на рис. 3-27.
3.3.3. Молекулы белков характеризуются чрезвычайным разнообразием [23]
Различия в природе боковых групп аминокислот обусловливает замечательное разнообразие возможных типов пространственной
структуры белков. Рассмотрим в качестве примера крайних случаев два типа белков, секретируемых клетками соединительной ткани, - коллаген и
эластин, которые относятся к белкам внеклеточного матрикса. В коллагене три отдельные полипептидные цепи, богатые пролином и содержащие в
каждом третьем положении глицин, закручены одна вокруг другой и образуют тройную спираль (см. разд. 14.2.6). Эти молекулы коллагена в свою
очередь упаковываются в волокна, в которых соседние молекулы скреплены ковалентными сшивками между соседними лизиновыми остатками. В
результате формируются волокна, способные выдерживать исключительно большую нагрузку (рис. 3-28).
Другой предельный случай - эластин, в котором относительно рыхлые и неструктурированные полипептидные цепи образуют благодаря
ковалентным сшивкам резиноподобную эластичную сеть, которая дает возможность таким тканям, как артерии и легкие, деформироваться и
растягиваться, не причиняя себе вреда. Как показано на рис. 3-29, эластичность обусловлена способностью индивидуальных молекул обратимо
разворачиваться под действием растягивающего усилия.
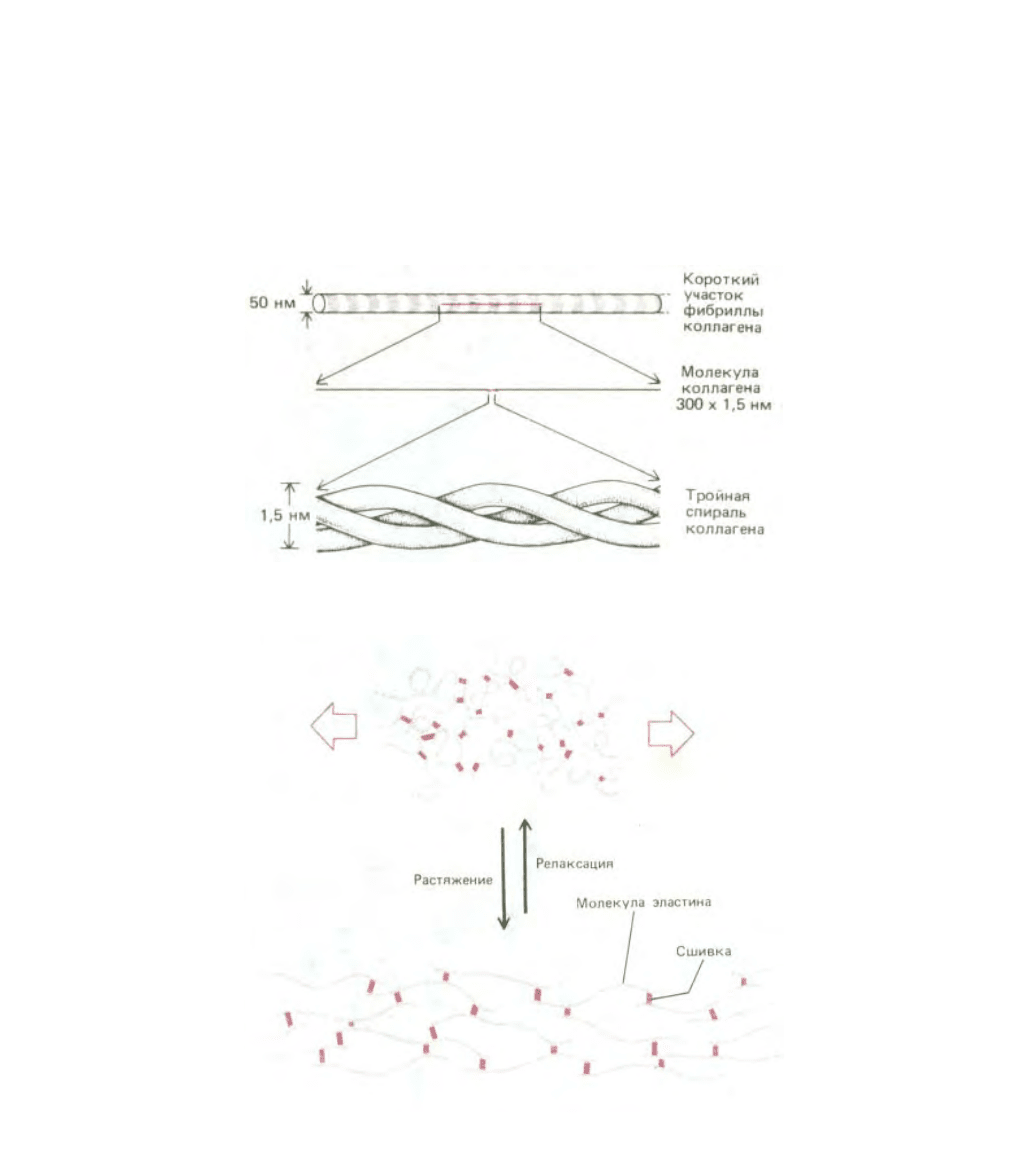
Рис. 3-28. Молекула коллагена - это тройная спираль, образованная тремя вытянутыми белковыми цепями. Множество сшитых вместе
стержнеобразных молекул коллагена образует прочные нерастяжимые коллагеновые фибриллы (вверху), которые обладают прочностью на
растяжение, сравнимой с прочностью стали.
Рис. 3-29. Эластин состоит из полипептидных цепей, образующих благодаря поперечным сшивкам, растяжимые волокна. При растяжении каждая
молекула эластина разворачивается, приобретая более протяженную конформацию. Разительный контраст между физическими свойствами
эластина и коллагена обусловлен большими различиями в их аминокислотных последовательностях.
143
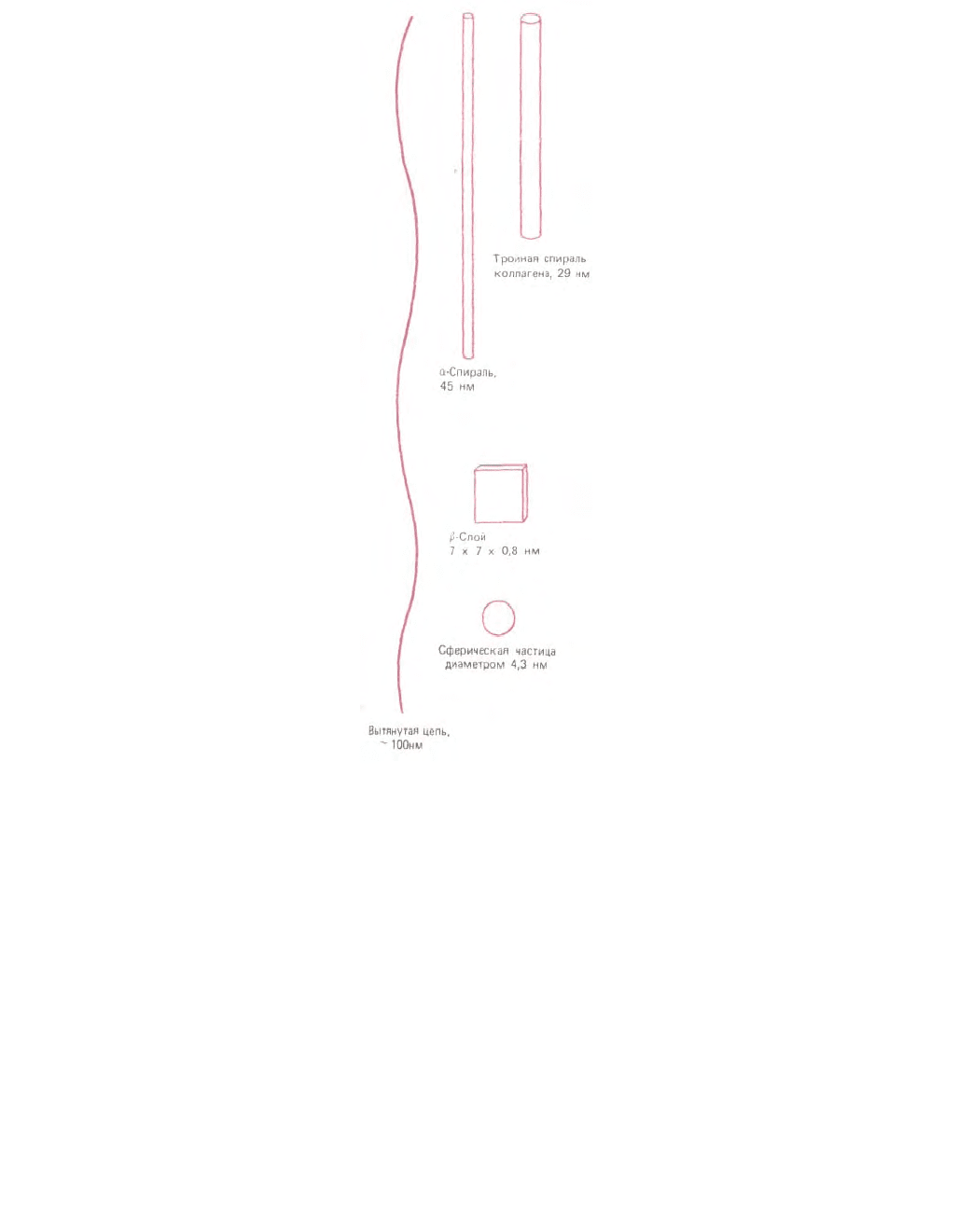
Рис. 3-30. Возможные размеры и форма молекулы белка из 300 аминокислот. Конкретная структура определяется последовательностью
аминокислот (D. Е. Metzler, Biochemistry, New York; Academic Press, 1977; печатается с изменениями).
Примечательно, что одна и та же химическая структура - аминокислотная цепь - может приобретать самую различную конформацию.
Назовем, например, резиноподобный эластин, похожий на стальной трос коллаген, разнообразные глобулярные белки - ферменты, очень
различающиеся по форме своей каталитической поверхности. На рис. 3-30 показано, сколь различную форму может в принципе принимать
полипептидная цепь длиной в 300 аминокислот. Реальная конформация, как мы уже отметили, полностью зависит от последовательности
аминокислот.
3.3.4. Белки имеют различные уровни пространственной организации [24]
Рассматривая структуру белка, полезно различать разные уровни его пространственной организации. Аминокислотную
последовательность называют первичной структурой белка. Регулярные водородные связи по всей длине непрерывной полипептидной цепи
приводят к образованию α-спиралей и β-слоев, которые представляют собой вторичную структуру белка. Некоторые комбинации α-спиралей и β-
слоев, упакованные вместе, формируют компактно уложенные глобулярные единицы, каждая из которых носит название белкового домена.
Домены обычно состоят из отрезков полипептидной цепи, содержащих от 50 до 350 аминокислот; по-видимому, они являются теми модульными
единицами, из которых строятся белки (см. ниже). Маленькие белки могут содержать только один домен, более крупные белки состоят из
нескольких доменов, связанных сравнительно открытыми участками полипептидной цепи. Наконец, отдельные полипептиды могут служить
субъединицами для формирования более крупных молекул, часто называемых белковыми агрегатами, или белковыми комплексами. В таких
комплексах субъединицы связаны друг с другом большим числом слабых нековалентных взаимодействий (см. разд. 3.1.1), у внеклеточных белков
эти взаимодействия часто стабилизированы дисульфидными связями.
Пространственную структуру белка можно проиллюстрировать несколькими способами. Рассмотрим, например, необычно маленький
белок - основной ингибитор трипсина поджелудочной железы, который содержит 58 аминокислотных остатков, упакованных в один домен. Этот
белок можно представить в виде стереопары, показывающей все его неводородные атомы (рис. 3-31, А), или в виде тщательно выполненной
трехмерной модели, где опущены многие детали (рис. 3-31, Б). Белок можно изобразить и более схематично, без боковых групп и атомов, чтобы
сфокусировать внимание на последовательности основной полипептидной цепи (рис. 3-31, В, Г и Д). Такие схематические представления очень
важны для выявления структуры белков, которые обычно крупнее, чем основной ингибитор трипсина, так как они дают возможность проследить за
нерегулярным расположением полипептидной цепи внутри каждого домена (рис. 3-32).
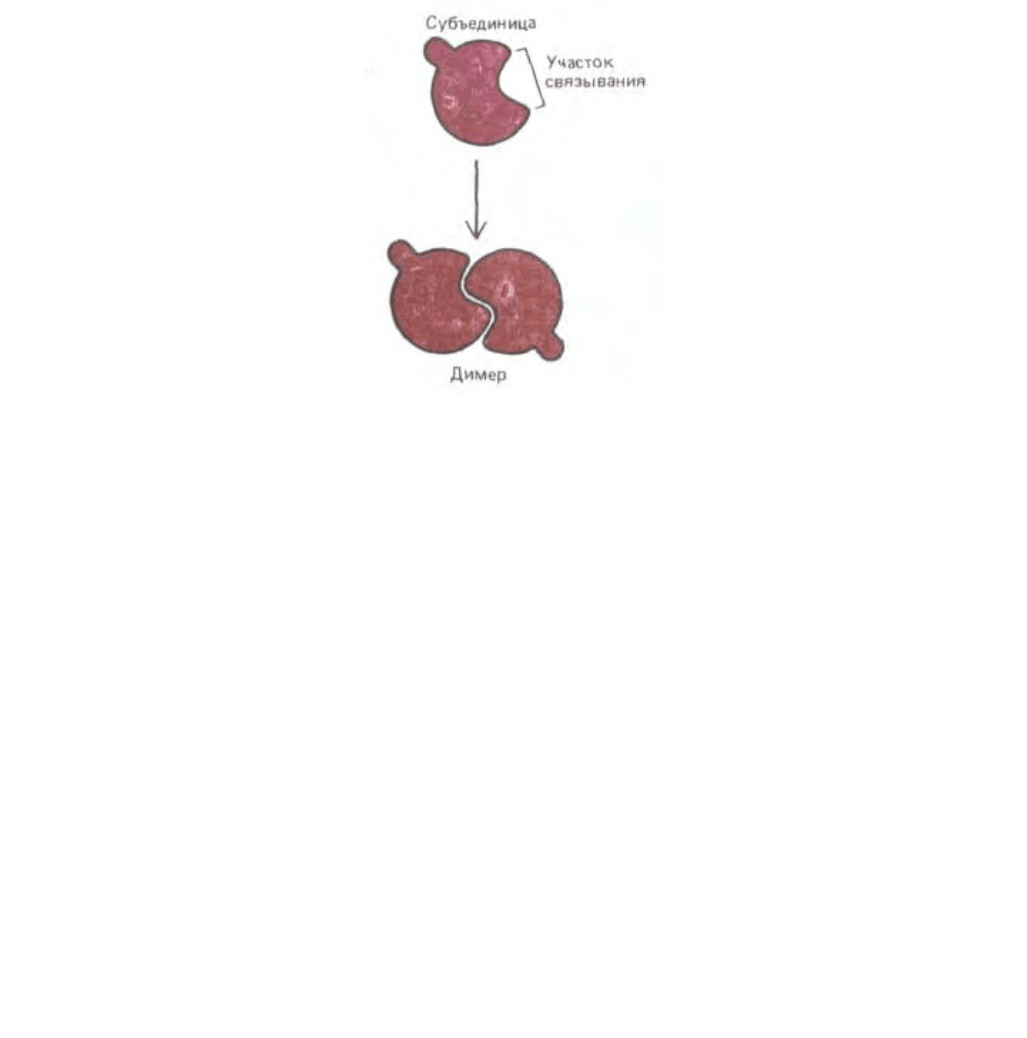
На рис. 3-33 показано, как структура большого белка может быть сведена к разным уровням организации, каждый из которых
иерархическим образом строится из предыдущих. Эти уровни возрастающей структурной организации, возможно, соответствуют стадиям
свертывания новосинтезированного белка в конечную нативную структуру внутри клетки.
144
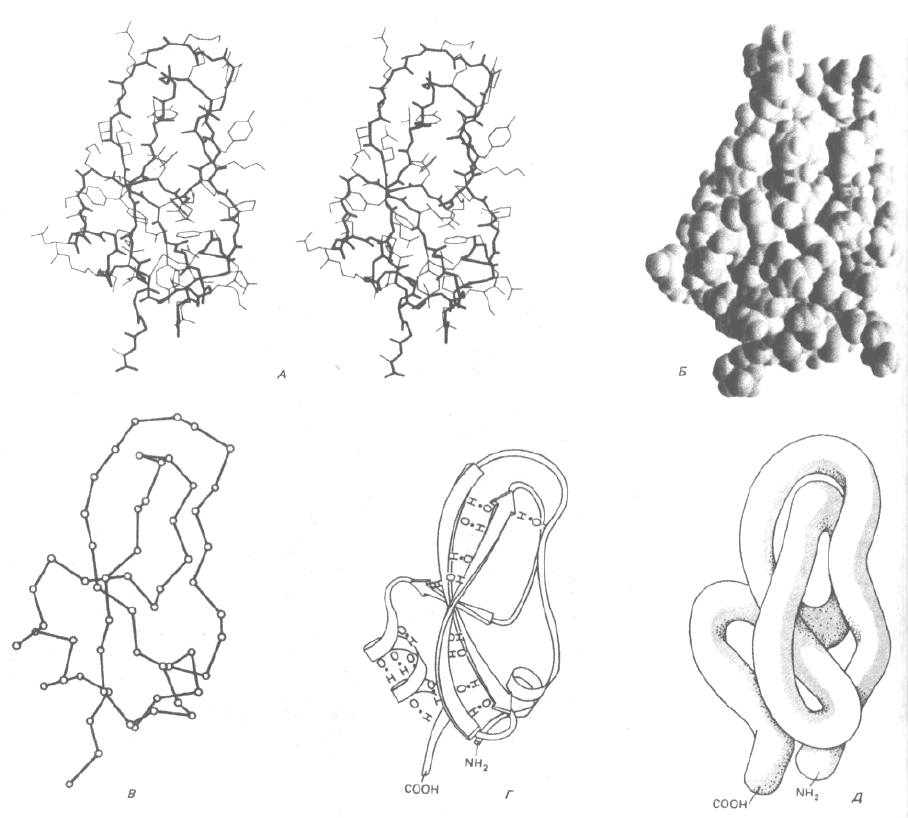
Рис. 3-31. Пространственная конформация малого белка основного ингибитора трипсина поджелудочной железы в пяти обычно использующихся
вариантах изображения. А. Стереопара, показывающая положение всех неводородных атомов. Основная цепь выделена жирной линией, а боковые
цепи - тонкими. Б. Пространственная модель, показывающая вандерваальсовы радиусы всех атомов (см. схему 3-1). В. Скелетная проволочная
модель, составленная из отрезков, соединяющих все атомы α-углерода вдоль полипептидного скелета. Г. Ленточная модель, которая представляет
все участки регулярных водородных связей либо в виде спиралей (α-спирали), либо в виде набора стрелок (β-слои), указывающих на карбоксил-
терминальный конец цепи; в этой модели также показаны водородные связи. Д. «Сосисочная» модель, которая демонстрирует расположение
полипептидной цепи без всяких деталей. Следует иметь в виду, что сердцевина всех глобулярных белков плотно заполнена атомами, и впечатление
пустого пространства вызвано только характером моделей В, Г и Д. (Б и В с любезного разрешения Richard J. Feldmann; А и Г - с любезного
разрешения Jane Richardson.)
145
Рис. 3-32. Ленточные модели пространственной структуры некоторых белковых доменов с разной организацией. А. Цитохром b
562
, однодоменный
белок, почти целиком состоящий из α-спиралей. Б. NAD-связанный домен лактат-дегидрогеназы, состоящий из смеси α-спиралей и β-слоев. В.
Изменчивый домен одной легкой цепи иммуноглобулина в виде сандвича из двух β-слоев. На этих рисунках α-спирали и соединительные цепи
окрашены, а цепи, составленные из β-слоев, изображены серыми стрелками. Обратите внимание, что полипептидная цепь, как правило, пересекает
домен два раза, делая резкие изгибы только на поверхности белковой молекулы. (Рисунок любезно предоставлен Jane Richardson.)
Рис. 3-33. Пространственная структура белка может быть описана в терминах различных уровней свертывания, каждый из которых составлен из
структур предшествующего уровня в иерархическом порядке. Такие уровни иллюстрируются на этом рисунке на примере двухдоменного
бактериального белка, активирующего катаболизм. Когда большой домен связывается с циклическим AMP, в белке происходит конформационное
изменение, дающее возможность малому домену связываться со специфической последовательностью ДНК. Аминокислотная последовательность
определяется как первичная структура белка, а первый уровень свертывания полипептидной цепи - как его вторичная структура. Как обозначено
внизу рисунка под квадратными скобками, комбинацию второго и третьего уровней свертывания, представленную здесь, обычно называют
третичной структурой, а четвертый уровень (комбинация субъединиц) - четвертичной структурой белка. (С изменениями с рисунков Jane
Richardson.)
146
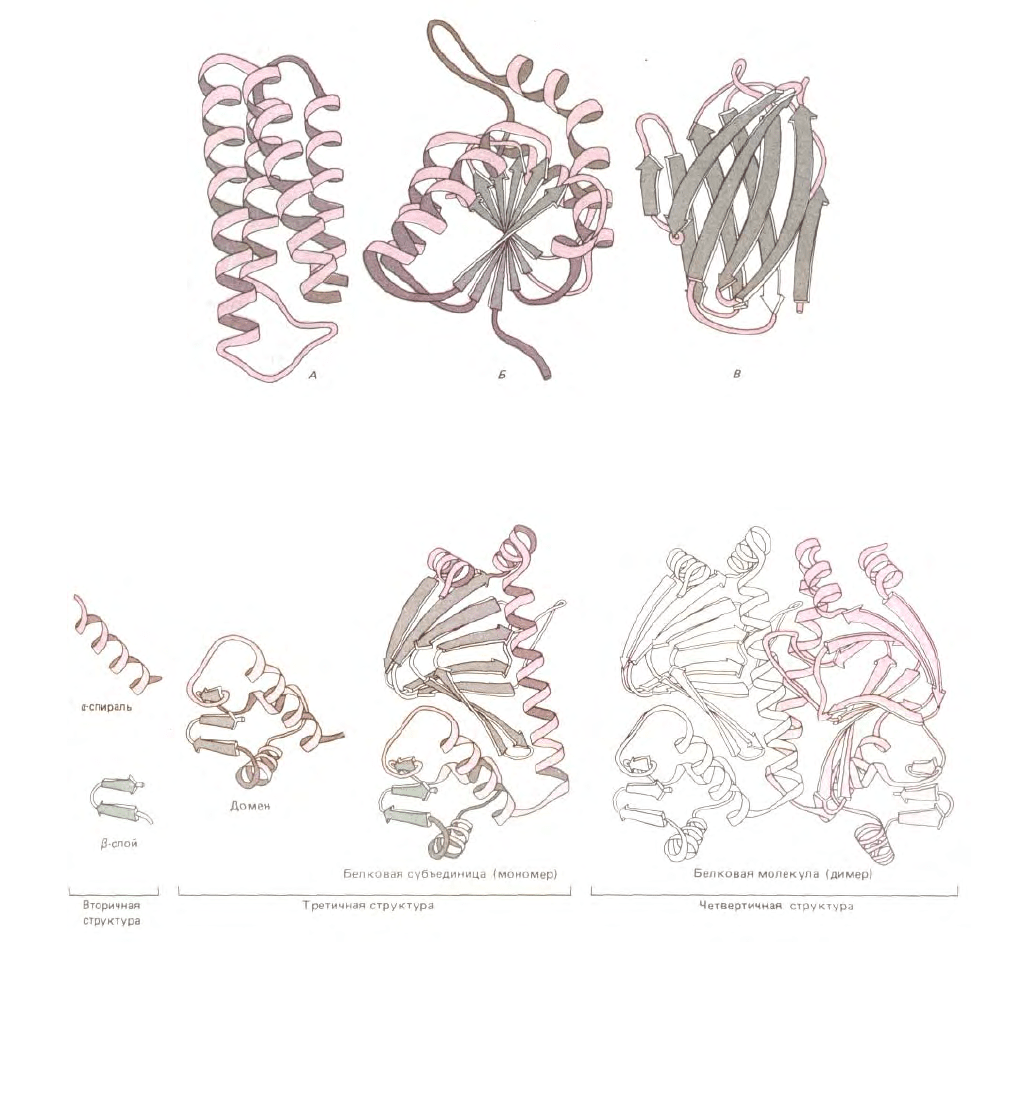
Рис. 3-34. Сопоставление аминокислотных последовательностей двух представителей семейства сериновых протеиназ. Показаны карбоксил-
терминальные участки двух белков (от 149 до 245-й аминокислоты). Одинаковые аминокислоты соединены цветными штрихами, а сериновые
остатки в активных центрах в положении 195 «высвечены». В участках полипептидных цепей, выделенных цветными прямоугольниками, каждая
аминокислота этих двух ферментов в трехмерной структуре занимает одинаковое положение (см. рис. 3-35). Б. Стандартные однобуквенные и
трехбуквенные обозначения аминокислот. (С изменениями из J. Greer, Proc. Natl, Acad. Sci. USA 77: 3393-3397, 1980.)
3.3.5. Сравнительно немногие потенциально возможные полипептидные цени могут оказаться полезными
Поскольку все 20 аминокислот химически различны и каждая может в принципе занимать в полипептидной цепи любое положение, то
для пептида из четырех аминокислот возможны 20ּ20ּ20ּ20 = 160000 различных цепей, а для полипептида из п аминокислот – 20
n
цепей. Таким
образом, может существовать более 10
390
различных белков со средней типичной длиной около 300 аминокислот.
Мы, однако, знаем, что лишь очень небольшая часть всех возможных белков примет стабильную пространственную конформацию. Все
остальные должны иметь множество различных конформаций с разными химическими свойствами и приблизительно одинаковой энергией. Белки с
такими изменчивыми свойствами не могут быть полезными и, следовательно, должны устраняться естественным отбором в ходе эволюции.
Удивительно точная пригнанность структуры современных белков к выполняемой ими функции обеспечивается их способностью
свертываться уникальным образом. Последовательность аминокислот не только обеспечивает исключительную стабильность одной из
конформаций, но и определяет необходимые для выполнения в клетке каталитической или структурной функции особенности этой конформаций и
ее химические свойства. Белки строятся настолько точно, что замена даже нескольких атомов одной аминокислоты может нарушить структуру и
привести к катастрофическим изменениям функции.
3.3.6. Новые белки часто возникают в результате незначительных изменений старых [25]
У клетки есть генетические механизмы, обеспечивающие дупликацию, модификацию и рекомбинацию генов в процессе эволюции (см,
разд. 10.5.1). Следовательно, если уже какой-нибудь белок с полезными свойствами поверхности раз возникнет, то его основная структура может
затем войти в состав многих других белков. В современных организмах различные белки с родственными функциями часто имеют схожую
последовательность аминокислот. Считается, что такие семейства белков возникли путем дупликации одного предкового гена и последующего
накопления в эволюции мутаций, постепенно обусловивших появление родственных белков с новыми функциями.
147
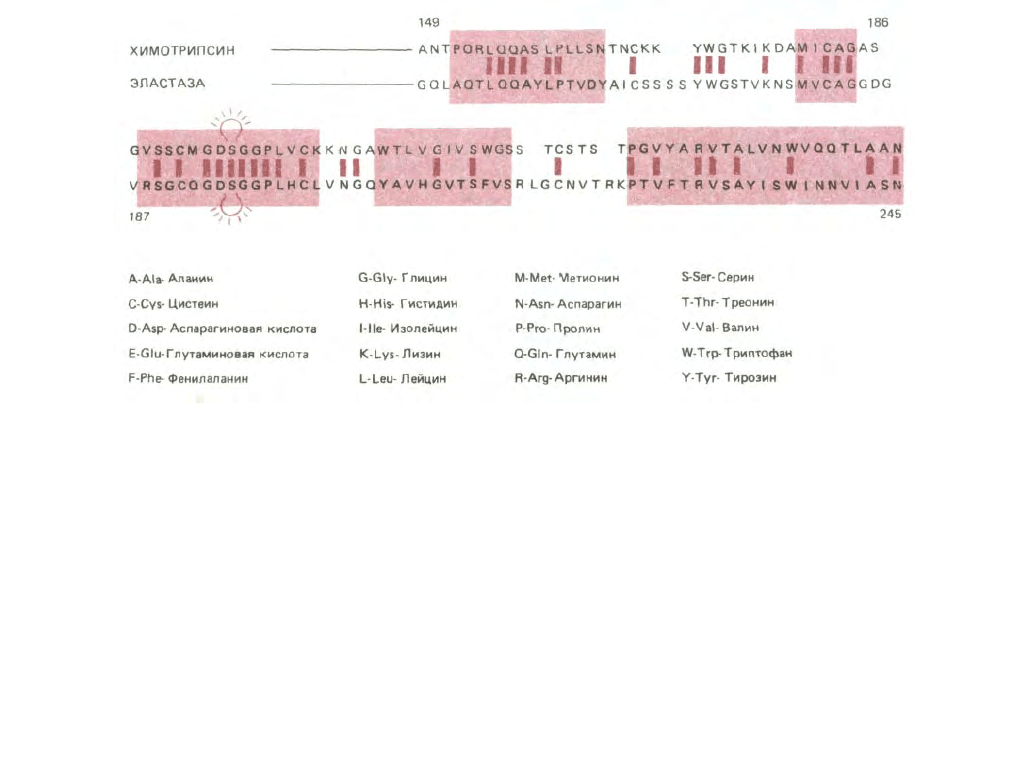
Рис. 3-35. Сравнение пространственной структуры эластазы (А) и химотрипсина (Б). У этих эволюционно родственных протеиназ одинаковы лишь
те аминокислоты, которые расположены в выделенных цветом участках полипептидной цепи. Тем не менее конформации белков очень похожи.
Обведены активные центры ферментов; оба активных центра содержат активированный остаток серина (см. рис. 3-47). Молекула химотрипсина
имеет несколько (более двух) концов цепи, поскольку она образована протеолитическим расщеплением химотрипсиногена, неактивного
предшественника.
Рассмотрим семейство протеолитических (расщепляющих) ферментов, сериновые протеиназы, включающие в себя пищеварительные
ферменты химотрипсин, трипсин и эластазу, а также многие из факторов свертывания - протеиназ, контролирующих процесс свертывания крови.
При сравнении любых двух ферментов этого семейства оказывается, что примерно 40% положений в полипептидной цепи занимают одни и те же
аминокислоты (рис. 3-34). Еще более поразительное сходство выявляется при сравнении их конформаций, определенных методом
рентгеноструктурного анализа: большинство поворотов и изгибов полипептидных цепей длиной в несколько сот аминокислот оказываются
идентичными (рис. 3-35).
Тем не менее разные сериновые протеиназы имеют совершенно различные функции. Некоторые из аминокислотных замен,
обусловивших различия ферментов этой группы, по-видимому, были отобраны в процессе эволюции, потому что привели к изменениям
субстратной специфичности и регуляторных свойств белков, что в свою очередь породило все многообразие современных функциональных
свойств. Другие аминокислотные замены могли быть «нейтральными», т. е. сохранились, потому что не повлияли ни на структуру, ни на функции
белка. Поскольку мутирование - процесс случайный, должны были происходить и вредные замены, изменяющие пространственную структуру
фермента достаточно сильно, чтобы его инактивировать. Эти измененные варианты были потеряны в процессе эволюции, так как производившие
их индивидуальные организмы должны были оказаться в невыгодных условиях и исчезнуть в результате естественного отбора. Поэтому
совершенно неудивительно, что клетки содержат целый набор структурно родственных полипептидных цепей, имеющих общих предков, но
выполняющих разные функции.

148
3.3.7. Новые белки часто возникают в результате объединения разных полипептидных доменов [26]
При возникновении в клетке ряда стабильных белковых поверхностей новые поверхности с иной специфичностью связывания могут
создаваться в результате объединения двух или более индивидуальных белков путем нековалентных взаимодействий. Для клеток характерно такое
объединение глобулярных белков в более крупные функциональные белковые агрегаты: молекулярная масса многих белковых агрегатов достигает
1 млн. и более, хотя молекулярная масса типичной полипептидной цепи составляет всего лишь 40000-50000 (приблизительно 300-400
аминокислот); размер лишь немногих полипептидов втрое превышает эту среднюю величину.
Сходный, но другой способ образования новых белков из существующих полипептидных цепей - это слияние соответствующих
последовательностей ДНК таким образом, что образуется ген, кодирующий одну большую полипептидную цепь (см. разд. 10.5.4). Считается,
что белки, возникшие этим путем, в разных частях полипептидной цепи свертываются независимо в отдельные глобулярные домены. Такая
«мультидоменная» структура характерна для многих белков, и, как
Рис. 3-36. Общий принцип, по которому наложение двух различных белковых поверхностей в процессе эволюции, приводит к появлению белков,
которые содержат новые центры связывания для других молекул. Как показано на этом рисунке, лиганд - связывающие центры часто расположены
в месте соприкосновения двух белковых доменов.
Рис. 3-37. Структура гликолитического фермента глицеральдегид-3-фосфат- дегидрогеназы. Белок состоит из двух доменов (выделены разным
цветом). Участки α-спирали представлены в виде цилиндров, а β-слоев- стрелками. Реакция, катализируемая этим ферментом, подробно приведена
на рис. 2-21. Заметим, что три центра связывания субстратов расположены в зоне соприкосновения двух доменов. (С любезного разрешения Alan. J.
Wonacott.)

149
Рис. 3-38. Пример широко распространенной в эволюции белков «перетасовки» блоков белковых последовательностей. Участки белка,
обозначенные окрашенными геометрическими фигурами, являются эволюционно родственными, но не идентичными. А. Бактериальный САР-белок
состоит из двух доменов; один из них (закрашенный треугольник) связывается со специфической последовательностью ДНК, второй - связывает
сАМР (см. рис. 3-33). ДНК-связывающий домен родствен ДНК-связывающим доменам многих других белков регуляторных генов, включая белки
lac-репрессор и erо-репрессор. Кроме того, две копии сАРМ-связывающего домена обнаружены в эукариотических киназах, регулируемых
связыванием циклических нуклеотидов. Б. Представлены два домена, состоящие примерно из 40 аминокислот, каждый из которых встречается в
трех больших белках позвоночных. Например, рецептор липопротеина низкой плотности (ЛНП) - это трансмембранный белок из 839
аминокислотных остатков, ответственный за выведение холестерола из клеток. Он содержит много доменов, имеющихся и в других белках, в
частности, семь копий цистеин - богатого домена (светлые кружки), участвующих в связывании ЛНП, и три копии такого же размера (окрашенные
кружки), функции которых неизвестны.
и следовало ожидать, исходя из рассмотренных выше эволюционных предпосылок, функционально важные центры связывания часто оказываются
расположенными на границе разных доменов (рис. 3-36). На рис. 3-37 показана структура конкретного мультидоменного белка.
Другой путь повторного использования аминокислотной последовательности особенно распространен среди длинных фибриллярных
белков, таких, как коллаген (см. рис. 3-28). В этом случае их структура формируется из многократных внутренних повторов предковой
аминокислотной последовательности. Ясно, что сведение вместе аминокислотных последовательностей путем объединения ранее существовавших
кодирующих последовательностей ДНК, является более эффективной стратегией для клетки, чем получать новые белковые последовательности в
результате случайных мутаций ДНК.
3.3.8. Структурные гомологии могут помочь в определении функций вновь открытых белков [27]
Развитие методов быстрого секвенирования молекул ДНК сделало возможным определение аминокислотных последовательностей
многих белков и нуклеотидных последовательностей соответствующих генов (см. разд. 4.6.6). Постоянно пополняемая «база данных белков»
обрабатывается на компьютере для поиска возможных гомологии последовательностей между вновь секвенированным белком и изученными ранее.
В настоящее время определена последовательность небольшого числа белков эукариотических организмов, при этом часто оказывается, что вновь
секвенированный белок является гомологом уже известного белка в пределах какого-то участка его длины. Отсюда следует, что большинство
белков, видимо, произошло от ограниченного числа предковых типов. Как и предполагалось, в последовательностях многих больших белков часто
видны признаки того, что они возникли путем объединения ранее существовавших доменов в новых комбинациях, так называемого процесса
«тасования доменов» (рис. 3-38).
Установление гомологии доменов также может быть полезным в другом аспекте. Определить пространственную структуру белка
намного труднее, чем определить аминокислотную последовательность. Однако конфигурация домена вновь секвенированного белка может быть
«отгадана», если он гомологичен домену белка, конформация которого ранее была определена методом рентгеноструктурного анализа. Часто
можно с приемлемой точностью определить структуру нового белка, предполагая, что повороты и изгибы полипептидной цепи в двух белках будут
одинаковыми, даже если есть отличия в аминокислотной последовательности.
150
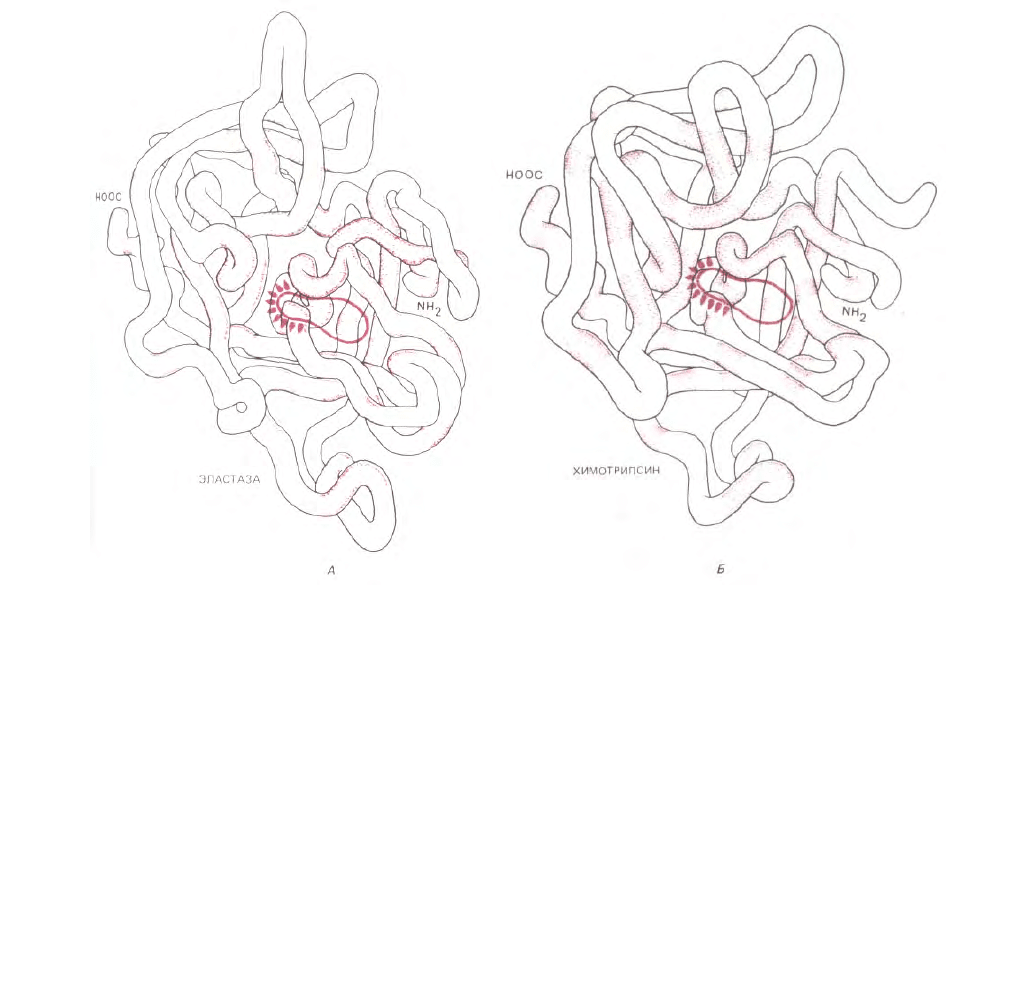
Рис. 3-39. Схема образования димера из идентичных белковых субъединиц. Если центр связывания узнает сам себя, димеры будут симметричными.
Эти пары часто в дальнейшем объединяются с другими субъединицами с образованием тетрамеров и более сложных ансамблей.
Такие сравнения белков важны еще и в том отношении, что сходные структуры часто предполагают и сходные функции. Можно
избежать многолетних экспериментальных исследований, установив гомологию аминокислотной последовательности с белком, функция которого
известна. Например, такие гомологии последовательностей впервые указали на то, что некоторые регуляторные гены клеточного цикла дрожжей и
некоторые гены, вызывающие раковое перерождение клеток млекопитающих, кодируют протеинкиназы. Таким же способом было определено, что
многие из белков, контролирующих морфогенез у плодовой мушки Drosophila, являются белками регуляторного гена, а один белок, участвующий в
морфогенезе, был идентифицирован как сериновая протеиназа.
Каждый год эта база данных пополняется все новыми сведениями о белковых последовательностях, что увеличивает вероятность
обнаружения полезных гомологии. Таким образом, сравнение аминокислотных последовательностей белков будет становиться все более важным
инструментом клеточной биологии.
3.3.9. Белковые субъединицы способны к самосборке в большие клеточные структуры [28]
Принцип, позволяющий белковым доменам ассоциировать с образованием новых центров связывания, «работает» и при сборке
значительно более крупных клеточных структур. Надмолекулярные структуры, такие, как ферментные комплексы, рибосомы, белковые волокна,
вирусы и мембраны, не синтезируются в виде единых гигантских молекул, связанных ковалентными взаимодействиями, а собираются в результате
нековалентной агрегации макромолекулярных субъединиц.
Использование субъединиц для построения больших структур имеет несколько преимуществ: 1) для построения большой структуры из
многократно повторенных субъединиц меньшего размера требуется меньше генетической информации; 2) поскольку субъединицы связаны между
собой многими сравнительно слабыми связями, их сборка и диссоциация легко поддаются контролю; 3) сборка структуры из субъединиц позволяет
сводить к минимуму количество ошибок, так как функционирование специального механизма корректирования в процессе сборки может устранять
испорченные субъединицы.
3.3.10. Одинаковые белковые субъединицы могут взаимодействовать с образованием геометрически регулярных
структур [29]
При наличии в белке центра связывания, комплементарного какому-либо участку на его собственной поверхности, белок будет
самопроизвольно агрегировать. В простейшем случае центр связывания узнает сам себя, и в результате образуется симметричный димер. Многие
ферменты и другие белки образуют такие димеры, которые часто в свою очередь служат субъединицами для формирования более крупных
агрегатов (рис. 3-39 и рис. 3-40).
Если центр связывания белка комплементарен другому участку на своей поверхности, то образуется цепь субъединиц. При некоторых
взаимных ориентациях двух участков связывания цепь замкнется сама на себя и рост прекратится. В результате образуется кольцо из двух, трех,
четырех или большего числа субъединиц (рис. 3-41). В более общем случае получится бесконечно длинный полимер из белковых субъединиц. При
условии, что все субъединицы связаны друг с другом идентичным