Moss Tom. DNA-protein interactions: principles and protocols
Подождите немного. Документ загружается.

EMSAs for Analysis of DNA–Protein 15
1.2. Applications of the EMSA
Because EMSA often allows the detection of specific DNA-binding pro-
teins in unpurified protein extracts (see ref. 9 and Fig. 1A), the technique has
been widely used to analyze crude cell or tissue extracts or partially purified
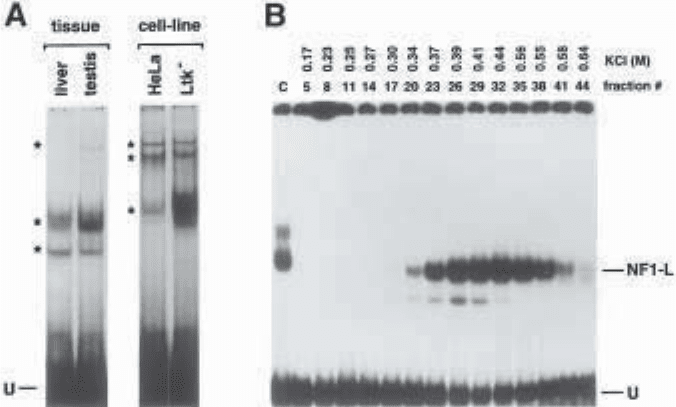
Fig. 1. Panel A. Autoradiograph of an EMSA performed using crude nuclear pro-
teins from both whole rat tissues and established tissue-culture cells. A 33-bp syn-
thetic oligonucleotide bearing the DNA sequence from the initiator site of the rat PARP
gene promoter was 5' end-labeled and used as a probe in EMSA. It was incubated with
crude nuclear proteins (5 µg) obtained either from fresh rat tissues (liver and testis) or
from established tissue-culture cells (HeLa and Ltk
–
). A number of nuclear proteins
(indicated by asterisks) were found to bind the rPARP promoter with varying efficien-
cies and most were common to both the tissues and the cell lines selected. U: Unbound
fraction of the labeled probe.
Panel B. Monitoring the enrichment of a nuclear protein by EMSA. Crude nuclear
proteins (50 mg) of a rat liver extract were prepared and further purified on a heparin–
Sepharose column. Nuclear proteins were eluted using a 0.1–1.0 M KCl gradient and
fractions individually incubated with a 34-bp double-stranded synthetic oligonucle-
otide bearing the DNA sequence of the rat growth hormone promoter proximal
silencer-1 element as the labeled probe. Both the concentration of KCl required to
elute the proteins contained in each fraction, as well as the fraction number selected
are indicated, along with the position of a major shifted DNA–protein complex corre-
sponding to the rat liver form of the transcription factor NF1 (termed NF1-L). C: con-
trol lane in which the silencer-1 labeled probe was incubated with 5 µg crude nuclear
proteins from rat liver; U: unbound fraction of the labeled probe.
16 Laniel, Béliveau, and Guérin
extracts for the presence of protein factors implicated in transcription (10–13)
and in DNA replication (9,14), recombination (15), and repair (16). The use of
unlabeled competitor DNA fragments further aids in identification of DNA-
binding proteins (see ref. 9, 15, and 17 and Fig. 2A), and their purification can
be easily monitored by EMSA (see ref. 9, and 13 and Fig. 1B). Moreover,
mutation or bases delection on the labeled DNA probe is often an efficient
approach to use when identifying the binding site of the protein of interest
(10,12).
EMSA yields invaluable data when purified or recombinant proteins are to
be analyzed, because quantification and kinetic studies are rapidly achieved
(10,14). Parameters of a DNA–protein interaction, such as association, disso-
ciation, and affinity constants, can be accurately measured (2,3,7,10), and the
effect of salt, divalent metals, protein concentration and the temperature of
incubation on complex formation can be directly observed (see ref. 15, 20, and
21 and Fig. 3A,B). EMSA has also greatly contributed to the elaboration of
models of complex assembly in the areas of transcription (11), DNA replica-
tion (14) and DNA repair (16).
Although EMSA is an informative and versatile method on its own, it
becomes more powerful when used in combination with other techniques.
Methylation (23) and other forms of binding interference studies (see Chapters
14 to 16), where a partially modified DNA probe is used, help to define the
exact position of the DNA binding site of the protein (10,24). Immunological
methods using specific antibodies, as in supershift experiments (see refs. 12
and 13 and Fig. 2B), are also very helpful in identifying the identity of the
protein component of given complexes. However, when analyzing large or
multiprotein complexes, supershifts may not be suitable because the supershifted
complexes may not be distinguished from the shifted ones or may not identify
the different proteins involved. Immunoblotting of EMSA gels (25), “Shift-
Western blotting” (26) and immunodepletion EMSA (27) can be used to resolve
such problems. In addition, determination of the molecular weight of the DNA-
binding protein(s) identified by EMSA can be achieved by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), either directly (28)
or following ultraviolet cross-linking of the DNA–protein complex (29).
1.3. Overview of the Procedure
Several components are required for EMSA and may influence the outcome
of the procedure.
1.3.1. Nuclear Extract
The choice of protein extract is governed by the objective of the study.
Whole-cell or nuclear extracts are very useful in analyzing the regulatory
EMSAs for Analysis of DNA–Protein 17
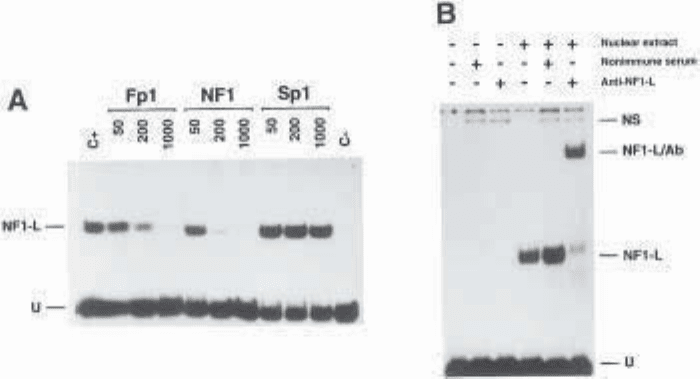
Fig. 2. Panel A. Competition in EMSA as a tool to evaluate the specific formation
of DNA–protein complexes. A synthetic double-stranded oligonucleotide bearing the
NF1 binding site from the Fp1 element of the human CRBP1 gene was 5' end labeled
and incubated with 1 µg of a heparin–Sepharose-enriched preparation of rat liver
NF1-L. Increasing concentrations (50-, 200-, and 1000-fold molar excess) of
unlabeled, double-stranded oligonucleotides containing various DNA binding sites
(Fp1, NF1, or Sp1) were added as competitors during the binding assays, and DNA/
protein complex formation was analyzed on native 8% polyacrylamide gels. Control
lanes containing the labeled probe alone (C–) or incubated with proteins in the
absence of any competitor DNA (C+) have also been included. The position of the
specifically retarded DNA/protein complex (NF1-L) and that of the free probe (U) is
also shown. (Modified from ref. 18: reprinted with permission from Mol. Endocrinol.,
Copyright [1994].)
Panel B. The identity of DNA-binding proteins as revealed by supershift analyses
in EMSA. The rGH silencer-1 labeled probe used in Fig. 1B was incubated with (+) or
without (–) 0.2 µg of a heparin–Sepharose-enriched preparation of NF1-L (see
panel A) , in the presence of either nonimmune serum (1 µL) or a polyclonal anti-
body directed against rat liver NF1-L. Formation of DNA/protein complexes was
then monitored by EMSA as in Fig. 1B. The position of the previously character-
ized NF1-L DNA/protein complex is shown (NF1-L) along with that of a
supershifted complex (NF1-L/Ab) resulting from the specific interaction of the
anti-NF1-L antibody with the NF1-L/silencer-1 complex. The position of a nonspe-
cific complex (NS), resulting from the binding of an unknown serum protein to the
labeled probe selected, is indicated, as well as the position of the remaining free probe
(U). (Modified from ref. 19: reprinted with permission from Eur. J. Biochem., Copy-
right [1994].)
18 Laniel, Béliveau, and Guérin
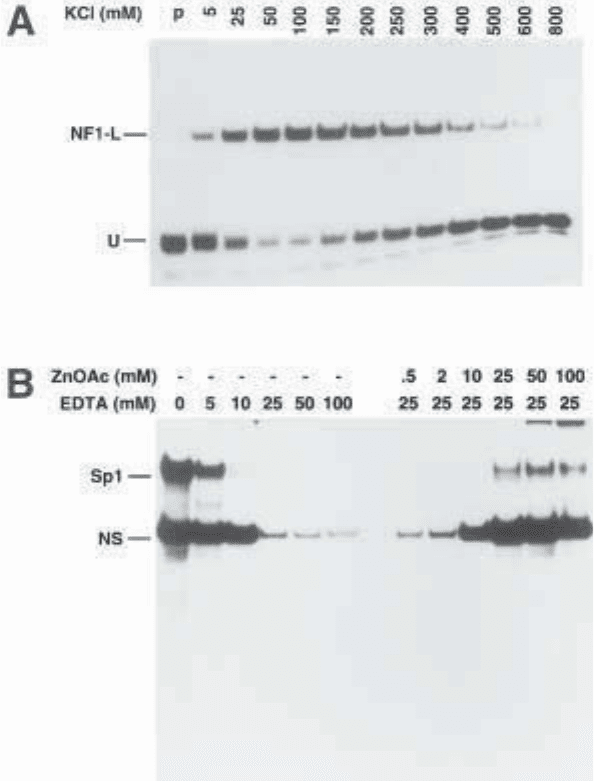
Fig. 3. Panel A. Salt-dependent formation of DNA–protein complexes in EMSA. A
5' end labeled 35-bp synthetic double-stranded oligonucleotide bearing the NF1-L
binding site of the 5'-flanking sequence of the human CRBP1 gene (and designated
Fp5) was incubated in the presence of 1 µg of a heparin–Sepharose-enriched prepara-
tion of NF1-L and increasing concentrations of KCl (5 to 800 mM) using binding
conditions similar to those described in this chapter. Formation of the Fp5/NF1-L
DNA–protein complex was then resolved by electrophoresis on a 4% native poly-
acrylamide gel. Very little free probe (U) is observed in the presence of either 50 or
100 mM KCl, providing evidence that optimal binding of NF1-L to its target site in
Fp5 is obtained at these salt concentrations. (Modified from ref. 20; reprinted with
permission from Biotechniques, Copyright [1992].)

EMSAs for Analysis of DNA–Protein 19
elements of a DNA fragment such as a gene promoter. Partial protein purifica-
tion allows further characterization of a DNA–protein interaction and can be
achieved by column chromatography on DNA-cellulose or heparin–Sepharose, or
by SDS-polyacrylamide gel fractionation and subsequent protein renaturation (see
ref. 30 and Note 1). Purified or recombinant proteins give valuable information on
protein interactions, competition, dimerization or cooperativity. Whatever pro-
tein extract used, its quality is a key factor in EMSA (see Notes 2 and 3).
1.3.2. DNA Probe
Cloned DNA fragments of 50–400 bp in length or synthetic oligonucleotides
of 20–70 nucleotides work very well in EMSA (see ref. 17 and Note 4) and
although double-stranded DNA is used most often, single-stranded DNA may
also be effective (15). Although larger DNA fragments usually encompass
more extensive regulatory sequences, oligonucleotides will generally contain
fewer protein binding sites and thereby yield more specific information,
the two approaches often complementing one another. The detection of
DNA–protein complexes is usually achieved by labeling of DNA probe (see
Note 5), and this is performed using a [
32
P]-labeled deoxynucleotide. However,
other, less hazardous methods are available (see Note 5), including labeling
with
33
P (31), with digoxygenin (32) or with biotin (33).
1.3.3. Gel Matrix
Acrylamide gels (see Note 6) combine high resolving power with broad
size-separation range and provide the most widely used matrix. Alternatively,
Panel B. DNA-binding properties of nuclear proteins revealed by EDTA chelation
in EMSA. A double-stranded synthetic oligonucleotide bearing the sequence of the rat
PARP US-1 binding site for the transcription activation factor Sp1 was 5' end-labeled
and incubated with 10 µg crude nuclear proteins from HeLa cells in the presence of
increasing concentrations of EDTA (0–100 mM) under binding conditions identical to
those described in this chapter. Formation of DNA/protein complexes was evaluated
by EMSA on a 8% polyacrylamide gel. As little as 10 mM EDTA proved to be sufficient to
chelate zinc ions and to totally prevent binding of Sp1 to the US-1 element. Similarly,
reaction mixtures containing the US-1 labeled probe incubated with 10 µg nuclear
proteins from HeLa cells in the presence of 25 mM final concentration of EDTA were
supplemented with increasing concentrations (0.5–100 mM) of zinc acetate (ZnOAc)
to evaluate the binding recovery for both Sp1 and the nonspecific DNA–protein complex
(NS). A substantial proportion of the DNA-binding capability of both the Sp1 and the NS
proteins could be recovered upon further addition of 25 mM zinc acetate, providing
evidence that both factors probably interact with DNA through the use of a Zn-finger-
containing DNA binding domain, a fact that was already known for Sp1. (Modified
from ref. 22: reprinted with permission from Eur. J. Biochem., Copyright [1993].)
20 Laniel, Béliveau, and Guérin
the use of less toxic, commercially available matrices has been reported (34–36).
Because of their larger pore size, agarose gels are sometimes used, either alone
or in combination with acrylamide, to study larger DNA fragments or
multiprotein complexes (37). Gel concentration is also important in EMSA
(see Note 7), however although lower concentration will generally allow the
resolution of larger complexes, it may affect their stability (7).
1.3.4. Buffer
Different low-ionic-strength buffers can be used in EMSA (see ref. 36 and
Note 8), and can include cofactors such as Mg
2+
or cAMP, which may be nec-
essary for some DNA–protein interactions (37).
1.3.5. Nonspecific Competitors
To ensure specificity of the DNA–protein interaction, a variety of nonspe-
cific competitors may be used. This is particularly important when using crude
protein extracts which contain nonspecific DNA-binding proteins. To avoid
nonspecific binding activities interfering with the EMSA, an excess of a non-
specific DNA such as salmon sperm DNA, calf thymus DNA or synthetic
DNAs such as poly(dI:dC) is used (see refs. 37 and 38 and Notes 9 and 10).
The addition of nonionic or zwitterionic detergents (39) or nonspecific pro-
teins (e.g., albumin [40]) may also increase specific DNA–protein interactions.
2. Materials
2.1. Probe Labeling
1. [γ
-32
P] ATP. Caution:
32
P emits high-energy beta radiation. Refer to the rules of
your local control radioactivity agency for handling and proper disposal of radio-
active materials and waste (see Note 5).
2. Approximately 25–50 ng of DNA from a 30-bp double-stranded oligonucleotide. For a
typical 70-bp probe derived from a subcloned promoter fragment, estimate the amount
of the plasmid DNA that is required to end up with about 100–200 ng of the DNA
fragment of interest following its isolation from the polyacrylamide gel (see Note 4).
3. Calf intestinal alkaline phosphatase (CIAP) and 10X CIAP reaction buffer: 0.5 M
Tris–HCl pH 9.0, 10 mM MgCl
2
, 1 mM ZnCl
2
, 10 mM spermidine.
4. T
4
polynucleotide kinase and 10X kinase buffer: 0.5 M Tris–HCl pH 7.5, 0.1 M
MgCl
2
, 40 mM DTT, 1 mM spermidine, 1 mM EDTA.
2.2. Probe Isolation
1. Standard electrophoresis apparatus for agarose gel.
2. Stock solution of 10X TBE: 0.89 M Tris, 0.89 M boric acid, and 20 mM EDTA.
3. 1% (w/v) agarose in 1X TBE supplemented with 0.5 µg/mL of ethidium bromide
from a 10-mg/mL solution. Caution: Ethidium bromide is a powerful mutagenic
agent (see Note 11).
EMSAs for Analysis of DNA–Protein 21
4. Restriction enzyme(s) with corresponding buffer(s).
5. For DNA precipitation, a preparation of 1 mg/mL tRNA, a solution of 3 M NaOAc
(pH 5.2), and a supply of dry ice.
6. Phenol/chloroform: Phenol saturated with 100 mM Tris–HCl pH 8.0.
7. 40% (w/v) 29:1 acrylamide–bisacrylamide: 29:1 (w/w) acrylamide and
N',N'-methylene bis-acrylamide. After complete dissolution of the components,
the solution should be filtered using Whatman No. 1 paper and can be stored at
room temperature. Caution: Acrylamide is a potent neurotoxic agent (see Note 6).
8. Dialysis tubing: molecular weight cutoff of 3500 and flat width of 18 mm.
9. Plastic wrap.
10. Autoradiography cassettes and film: Kodak XOmat AR.
2.3. Electrophoretic Mobility Shift Assay
1. Standard vertical electrophoresis apparatus for polyacrylamide gels, a gel length
of 15 cm is adequate. (See Note 12.)
2. 40% (w/v) 39:1 acrylamide–bisacrylamide: 39:1 (w/w) acrylamide and N',N'-methyl-
ene bis-acrylamide. Caution: Acrylamide is a potent neurotoxic agent (see Note 6).
3. 5X Tris–glycine: 250 mM Tris, 12,5 mM EDTA, and 2 M glycine. (See Note 8.)
4. Extract (crude or enriched) containing cell or tissue nuclear proteins. (See Note 2.)
5. 2X binding buffer: 20 mM HEPES pH 7.9, 20% glycerol, 0.2 mM EDTA, 1 mM
tetrasodium pyrophosphate (see Note 3) and 0.5 mM PMSF.
6. 6X loading buffer: 0.25% bromophenol blue, 0.25% xylene cyanol, and
40% sucrose.
7. Whatman chromatographic paper (3MM) and plastic wrap.
8. Standard gel dryer.
9. Autoradiography cassettes and film: Kodak XOmat AR.
3. Methods
3.1. Probe Labeling
3.1.1. Labeling DNA Fragments Derived from a Subcloned Sequence
1. Select restriction enzymes that produce the shortest DNA fragment containing
the sequence of interest. One of these restriction enzymes should produce a pro-
truding 5' end or blunt end to support labeling with T
4
polynucleotide kinase (see
Note 13). Following the manufacturer’s optimal enzymatic conditions, prepare a
digestion mix with one of the restriction enzymes in 50 µL to linearize the vector.
The initial amount of DNA should be calculated to end up with at least 100–200 ng
of DNA after double-restriction enzyme digestion and further isolation of the
DNA fragment from the polyacrylamide gel.
2. Before proceeding with dephosphorylation, make sure that digestion is complete
by loading a sample (50–100 ng) on a 1% (w/v) agarose minigel. Once complete
digestion of the plasmid DNA has been verified, add directly to the digestion
reaction mix 1 U of CIP, 10 µL of 10X CIP buffer, and fill to 100 µL with H
2
O.
Incubate at 37°C for 90 min.
22 Laniel, Béliveau, and Guérin
3. To totally eliminate and inactivate CIAP, transfer the reaction mix at 70°C for 10 min
and perform a phenol/chloroform followed by a chloroform extraction. Precipi-
tate DNA by adding a 1/10th volume of 3 M NaOAc, pH 5.2 and 2 volumes of
cold 95% ethanol. Allow DNA to precipitate on dry ice for 30 min, then centri-
fuge for 15 min.
4. Resuspend DNA in 33 µL of H
2
O, add 5 µL of 10X kinase buffer, 10 µL (100 µCi) of
[γ
-32
P]ATP and 2 µL of T
4
polynucleotide kinase. Mix and incubate at 37°C for 2 h.
5. Following the labeling procedure, reprecipitate DNA and resuspend in 30 µL of
H
2
O. Keep a 2-µL sample and digest the remainder with the second restriction
enzyme, following manufacturer’s conditions.
3.1.2. Labeling Double-Stranded Synthetic Oligonucleotides
1. Mix equal amounts of the complementary strands, heat at 5°C over the specific
melting temperature (T
M
) of the sequence for 5 min, and let cool to room tem-
perature (RT). When DNA reaches RT, place at 4°C for a few hours prior to use.
2. Use 25–50 ng of the double-stranded oligonucleotide preparation and perform
DNA labeling with T
4
polynucleotide kinase as described in step 4 of Subhead-
ing 3.1.1. but using 30 µCi of [γ
-32
P] ATP.
3.2. Probe Isolation
3.2.1. For a Typical 70-bp Probe Derived
from a Subcloned Promoter Fragment
1. Rigorously clean and dry the polyacrylamide gel apparatus and its accessories
prior to use. Gel plates should be cleaned using any good quality commercial
soap and then rinsed with 95% ethanol. One plate can be treated with a coat of
Sigmacote (chlorinated organopolysiloxane in heptane) to facilitate gel removal
from the plates after running.
2. Prepare a 6% polyacrylamide gel (41) as follows; mix 2.5 mL of 10X TBE, 3.75 mL
of 40% acrylamide (29:1) stock solution, and H
2
O to 25 mL final volume.
Add 180 µL of 10% ammonium persulfate and 30 µL of TEMED. Carefully
stir and pour the acrylamide solution between the plates. Insert well-forming
comb and allow the gel to set for 30 min., then mount the gel in the electro-
phoresis tank and fill the chamber with 1X TBE .
3. To the double-digested DNA, add 10 µL of 6X loading buffer and load into two
separate wells. For the 2-µL control sample from the single digestion, add 2 µL of
loading buffer and load in a free well. Migration should be stopped when bro-
mophenol blue, which is used as a migration marker, reaches two-thirds of
the gel length.
4. Carefully disassemble the apparatus and discard the running buffer as radio-
active waste. Remove one plate and leave the gel on the remaining plate. Cover
the gel with plastic wrap and, in a dark room, place a film over it. It is very
important to mark the exact position of the gel on the film as a reference. This
can be achieved by using [
32
P]-labeled black ink. Expose the film for 3 min
and develop.
EMSAs for Analysis of DNA–Protein 23
5. If the digestion step with restriction enzymes is complete, two labeled bands
resulting from the double digestion should appear on the autoradiogram (pro-
vided that each of the restriction enzymes selected initially cut the probe-bearing
recombinant plasmid only once). Using a razor blade, cut out from the film the
lower band corresponding to the selected probe. Replace the film on the gel
(which is still covered with plastic wrap), aligning the reference marks carefully.
Using the aperture in the film as guide, remove the probe-containing gel frag-
ment using a scalpel blade.
6. Place the acrylamide fragment in a dialysis tubing closed at one end and add
1 mL of 1X TBE. Remove any remaining air bubbles, close the other end, and
place the dialysis tubing in a standard horizontal electrophoresis tank filled with
1X TBE. Run at 100 V for 15 min.
7. Through the action of electrophoretic migration, the labeled probe will pass from
the acrylamide fragment to the TBE solution contained in the dialysis tubing.
DNA will concentrate as a thin line along the dialysis tubing (on the cathode
side) and must be removed by gently rubbing the tubing with a solid object. Using
a Pasteur pipet, transfer the labeled probe-containing TBE from the dialysis
tubing into three separate microcentrifuge tubes (about 300 µL each). Other pro-
cedures may also be selected for extracting the labeled probe from the polyacry-
lamide gel (42).
8. Repeat steps 6 and 7 to make sure that all of the probe has been eluted from the
acrylamide fragment. At the end of the second elution, recover the TBE again
into three other microcentrifuge tubes.
9. Precipitate the probe by adding 1/10th volume of 3 M NaOAc pH 5.2 and two vol-
umes of cold 95% ethanol. Allow labeled DNA to precipitate on dry ice for 30 min.
10. Centrifuge and discard the supernatant and resuspend DNA in 50 µL of sterile H
2
O.
Pool the samples into one microcentrifuge tube and reprecipitate as in step 9.
11. Estimate the recovery of labeled DNA by counting the Cerenkov radiation emit-
ted by the pellet using a β counter or by resuspending the DNA in a small volume
(100 µL) and counting a 1-µL aliquot in scintillation liquid.
12. Resuspend the labeled DNA in order to obtain 30,000 cpm/µL.
3.2.2. For a Double-Stranded Oligonucleotide Labeled Probe
Proceed as in Subheading 3.2.1. except that steps 1 through 8 should be
omitted and replaced by two sequential precipitations in the presence of 5 µg
total tRNA as described in step 9. (See Note 14.)
3.3. EMSA
1. Rigorously clean and dry the electrophoresis tank and its accessories prior to use
and treat the glass plates as previously described for probe isolation (step 1; Sub-
heading 3.2.1.).
2. For a typical 70-bp probe, prepare a 6% polyacrylamide gel (see Note 7) by mix-
ing 2.5 mL of 10X Tris–glycine, 3.75 mL of 40% acrylamide (39:1) stock solu-
tion, and H
2
O to 25 mL. Add 180 µL of 10% ammonium persulfate and 30 µL of
24 Laniel, Béliveau, and Guérin
TEMED. Carefully stir and pour the acrylamide solution between the plates (see
Note 15). Use a comb that has 0.8-cm-width teeth. Allow the gel to set for at least
2 h, then mount gel in the electrophoresis tank and fill the chamber with 1X Tris–
glycine (see Note 8). As soon as the gel is mounted and set, remove the comb and
carefully wash the wells with running buffer.
3. Prerun the gel at 4°C and 120 V (8 V/cm) until the current becomes invariant
(this takes around 30 min). Prerunning ensures that the gel will remain at a con-
stant temperature from the moment of sample loading.
4. When the gel is ready for loading, prepare samples as follows. For each sample,
mix 12 µL of 2X binding buffer, 1 µL of 1 mg/mL poly(dI:dC) (see Notes 9 and
10), and 0.6 µL of 2M KCl (see Note 10); then add 30,000 cpm of labeled probe.
Where possible, to minimize pipeting errors, prepare a single mix of the common
reaction components and distribute equal volumes into the reactions. Finally, add
1–10 µg protein extract and H
2
O to a final volume of 24 µL. Mix each tube gently
and incubate at RT for 3 min. As a control, prepare a sample without protein extract
and add 1 µL of 6X loading buffer containing bromophenol blue and xylene cyanol.
5. Load samples by changing the pipet tip for each sample.
6. Run at 120 V (8 V/cm) and let samples migrate until the free probe reaches the
bottom of the gel (see Note 16). In the case of a 70-bp probe loaded on 6%
acrylamide gel, this means 5–6 h of migration.
7. After the gel run, disassemble the apparatus and remove one of the glass plates,
place a Whatman paper over the gel, and carefully lift the gel off the remaining
plate. Make sure that the gel is well fixed on the Whatman before lifting the gel to
avoid gel breakage. Place plastic wrap over the gel and dry at 80°C for 30 min.
8. Place an X-ray film over the gel in an autoradiography cassette and expose at
–70°C overnight.
4. Notes
1. Very intense, large or smeary shifted complexes usually result from multiple
comigrating DNA–protein complexes that possess nearly identical electro-
phoretic mobilities in native polyacrylamide gels despite the fact that the pro-
teins they contain usually have distinctive molecular masses on denaturing
SDS-PAGE (43,44). An attractive method that helps to distinguish between the
proteins yielding these multiple, comigrating complexes is the SDS–polyacryla-
mide gel fractionation–renaturation procedure (30). This procedure allows
recovery and enrichment of specific proteins suitable for further analyzes by
EMSA, in addition to providing their approximate molecular masses.
2. When using crude nuclear extracts for detecting DNA–protein complexes in
EMSA, the quality of the extract is very critical. Whenever possible, nuclei puri-
fication procedures using a sucrose cushion or pad (45) is to be preferred in order
to eliminate contamination by cytosolic proteins that most often also contain
substantial amounts of proteases. Purifying nuclei on sucrose pads has generally
yielded high-quality nuclear extract samples. However, such extracts require
large quantities of fresh tissue, rendering the approach inappropriate when