Baeckvall J.-E. (ed.) Modern Oxidation Methods
Подождите немного. Документ загружается.


Table 1.4 Dihydroxylation of various alkenes with air.
a)
.
Entry Alkene Cat.
(mol%)
Ligand
(L)
L/Os [L]
(mmol/L)
Time
(h)
Yield
(%)
b)
Selectivity
(%)
b)
ee
(%)
1
0.5 (DHQD)
2
PHAL 3 : 1 3.0 24 42 42 87
2 0.5 (DHQD)
2
PHAL 3 : 1 3.0 16 66 66 86
3 0.5 (DHQD)
2
PHAL 3 : 1 3.0 14 76 76 87
4 0.5 (DHQD)
2
PHAL 3 : 1 3.0 24 88 88 89
5
O
0.5 (DHQD)
2
PHAL 3 : 1 3.0 24 63 63 67
6 0.5 (DHQD)
2
PHAL 3 : 1 3.0 18 68 68 68
7 0.5 (DHQD)
2
PHAL 3 : 1 3.0 14 67 67 66
8 0.5 (DHQD)
2
PHAL 3 : 1 3.0 9 77 77 68
9 0.5 ———24 0 (84) 0 (84) —
10
c)
1.0 DABCO 3 : 1 1.5 24 4 (77) 5 (87) —
11
c),d)
1.0 (DHQD)
2
PHAL 3.1 1.5 24 40 (35) 48 (42) 86
12
c),e)
1.0 (DHQD)
2
PHAL 3 : 1 1.5 24 89 (7) 89(7) 98
13
d)
C
4
H
9
C
4
H
9
1.0 (DHQD)
2
PHAL 3 : 1 6.0 24 85 85 82
(Continued)
1.2 Environmentally Friendly Terminal Oxidants
j
13

Table 1.4 (Continued )
Entry Alkene Cat.
(mol%)
Ligand
(L)
L/Os [L]
(mmol/L)
Time
(h)
Yield
(%)
b)
Selectivity
(%)
b)
ee
(%)
14
C
6
H
13
0.5 (DHQD)
2
PHAL 3 : 1 3.0 18 96 96 63
15 0.1 (DHQD)
2
PHAL 3 : 1 0.6 24 95 95 44
16 0.1 (DHQD)
2
PHAL 15 : 1 3.0 24 97 97 62
17
f)
0.1 (DHQD)
2
PHAL 3 : 1 1.5 24 94 94 47
18
f)
0.1 (DHQD)
2
PHAL 6 : 1 3.0 24 95 95 62
19
C
6
F
13
2.0 (DHQD)
2
PYR
g)
3 : 1 12.0 24 55 — 68
a) Reaction conditions: K
2
[OsO
2
(OH)
4
], 50
C, 2 mmol alkene, 20 atm air, pH ¼ 10.4, 25 mL buffer solution, 10 mL tert-BuOH, entries 9–12: 15 mL buffer solution, 20 mL
tert-BuOH, entries 17–18: 50 mL buffer solution, 20 mL tert-BuOH.
b) Values in brackets are for benzaldehyde.
c) 1 mmol alkene.
d) pH ¼ 12.
e) Isobutyl methyl ketone instead of tert-BuOH.
f) 10 mmol alkene.
g) Hydroquinidine 2,5-diphenyl-4,6-pyrimidinediyl diether.
14
j
1 Recent Developments in Metal-catalyzed Dihydroxylation of Alkenes
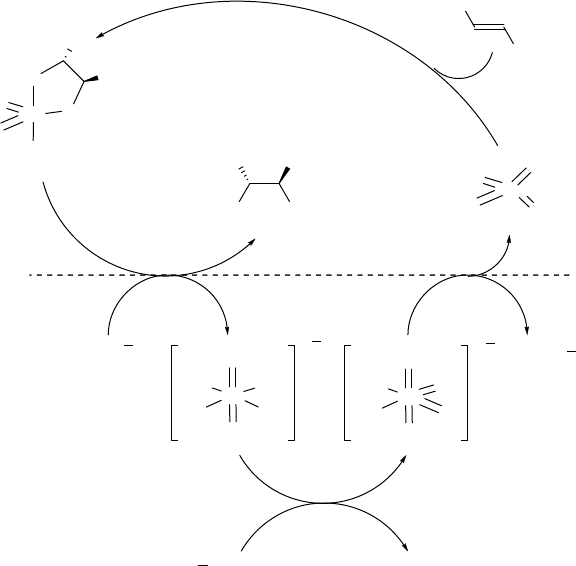
The mechanism of the dihydroxylation reaction with oxygen or air is thought to be
similar to the catalytic cycle presented by Sharpless et al. for the osmium-catalyzed
dihydroxylation with K
3
[Fe(CN)
6
] as the reoxidant (Scheme 1.11). The addition of
alkene to a ligated Os(VIII) species proceeds mainly in the organic phase. Depending
on the hydrolytic stability of the resulting Os(VI) glycolate complex, the rate-
determining step of the reaction is either hydrolysis of the Os(VI) glycolate or the
reoxidation of Os(VI) hydroxy species. There must be a minor involvement of a
second catalytic cycle, as suggested for the dihydroxylation with NMO [21]. Such a
second cycle would lead to significantly lower enantioselectivities, as the attack of
a second alkene molecule on the Os(VIII) glycolate would occur in the absence of
the chiral ligand. The observed enantioselectivities for the dihydroxylation with air
are only slightly lower than those in the data previously published by the Sharpless
group, despite the higher reaction temperature (50
Cvs0
C). Therefore the direct
oxidation of the Os(VI) glycolate to an Os(VIII) glycolate does not represent a major
reaction pathway.
R R
HO OH
Os
O
O
O
O
Os
O
O
O
O
L
Os
O
O
O
HO
O
HO
Os
OH
OH
O
HO
O
HO
2
2
L+
L+
OH2
H2
2
O
OH2
organic
aqueous
1
2
VIII
VI
VIII
VI
O
2
H
2
O
R
R
R
R
Scheme 1.11 Proposed catalytic cycle for the dihydroxylation of alkenes with OsO
4
and oxygen as
the terminal oxidant.
1.2 Environmentally Friendly Terminal Oxidants
j
15

1.3
Supported Osmium Catalyst
Hazardous toxicity and high costs are the chief drawbacks in reactions using osmium
tetroxide. However, these disadvantages can be overcome by the use of stable and
nonvolatile adducts of immobilized osmium tetroxide together with the advance-
ment of a regeneration procedure for osmium tetroxide from a stoichiometric
secondary oxidant, significantly improving the usability of osmium-catalyzed dihy-
droxylation [32]. These immobilized catalysts offer the advantages of easy and safe
handling, simple separation from the reaction medium, and the possibility to reuse
the expensive transition metal. Unfortunately, the instability of the support and
leaching of the metal continue to be the main drawbacks.
1.3.1
Nitrogen-group Donating Support
In this context Cainelli and coworkers reported in 1989 the preparation of polymer-
supported catalysts in which OsO
4
was immobilized on several amine-type poly-
mers [33]. Such catalysts have structures of the type OsO
4
L with the N-group of the
polymer (¼L) being coordinated to the Lewis acidic osmium center. Based upon this
concept, catalytic enantioselective dihydroxylation was established by using polymers
containing cinchona alkaloid derivatives [34]. However, since the amine ligands
coordinate to osmium under equilibrium conditions, recovery of the osmium using
polymer-supported ligands was difficult. In some cases, additional OsO
4
is needed to
maintain the reactivity. Os-diolate hydrolysis seems to require detachment from the
polymeric ligand, and hence causes leaching.
Herrmann and coworkers reported on the preparation of immobilized OsO
4
on
poly(4-vinyl pyridine) and its use in the dihydroxylation of alkenes by means of
hydrogen peroxide [35]. However, the problems of gradual polymer decomposition
and osmium leaching were not solved.
OsO
4
can also be immobilized in polyaniline through nitrogen coordination as
shown by Cloudarys group (Scheme 1.12) [36]. This polyaniline was applied to other
Lewis acid-type transition metals like Sc, Pd, Re, and In. An electron transfer
N
H
N
H
N N
OsO
4
N
H
N
H
N N
δ
-
OsO
4
OsO
4
OsO
4
OsO
4
δ
-
δ
+
δ
+
δ
+
δ
+
δ
-
δ
-
Scheme 1.12 Structure of PANI-doped with OsO
4
.
16
j
1 Recent Developments in Metal-catalyzed Dihydroxylation of Alkenes

mediator of MTO can be incorporated together with OsO
4
in this polyaniline to give a
multifunction supported catalyst, which catalyzes the asymmetric dihydroxylation of
trans-stilbene with 30% H
2
O
2
as the terminal oxidant (Scheme 1.13). This system
gave good isolated yields (81–85%) with excellent enantioselectivity (98% ee ) for 5-
times recycling.
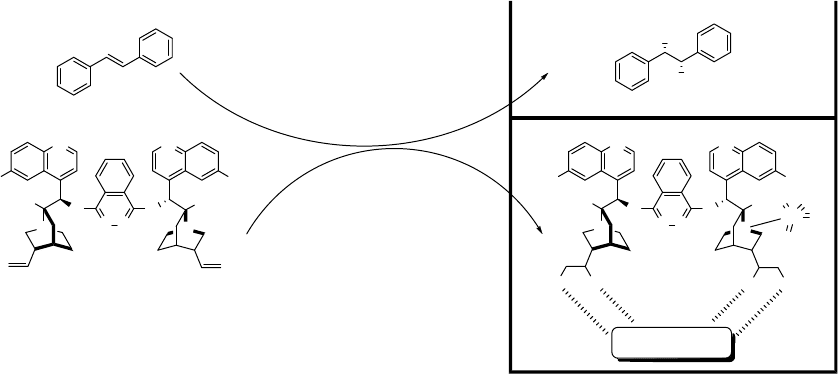
In another approach, Song and coworkers took advantage of the double bond in
naturally occurring quinine [37]. The dimeric ligand (QN)
2
PHAL is dihydroxylated
in situ. With hydrogen bonding interaction with sucrose in the aqueous phase, the
dihydroxylated ligand with coordinated OsO
4
stays in the aqueous phase in the
reaction mixture. Hence, dihydroxylation of trans-stilbene can be performed 4 times
in 82–92% yield and 81–99% ee with 0.1 mol% of OsO
4
and 2.5 mol% of (QN)
2
PHAL
(Scheme 1.14).
1.3.2
Microencapsulated OsO
4
A new strategy (at the time) of microencapsulated osmium tetroxide was published
by Kobayashi and coworkers in 1998 [38]. The metal is immobilized onto a polymer
on the basis of physical envelopment by the polymer and on electron interactions
between the p electrons of the benzene rings of the polystyrene-based polymer and a
vacant orbital of the Lewis acid. Using cyclohexene as a model compound it was
shown that this microencapsulated osmium tetroxide (MC OsO
4
) can be used as a
catalyst in the dihydroxylation with NMO as stoichiometric oxidant (Scheme 1.15).
In contrast to other typical OsO
4
-catalyzed dihydroxylations, where H
2
O-
t
BuOH
is used as solvent system, the best yields were obtained in H
2
O-acetone-CH
3
CN.
While the reaction was successfully carried out using NMO, moderate yields were
obtained using trimethylamine N-oxide, and much lower yields were observed using
hydrogen peroxide or potassium ferricyanide. The catalyst was recovered quantita-
tively by simple filtration and reused five times without any loss in activity.
A study on the rate of conversion of the starting material showed that the reaction
proceeds faster using OsO
4
than using the microencapsulated catalyst. This is
ascribed to the slower reoxidation of the microencapsulated osmium ester with
NMO, compared to simple OsO
4
.
Later on, acrylonitrile-butadiene-polystyrene (ABS) polymer was used as the
support based on the same microencapsulation technique, and several alkenes,
PANI-Os-Remol%2
(DHQD)mol%6
2
PHAL
H/acetone
2
0°CO,
Eteq2
4
NOAc
H30%eq1.5
2
O
2
yield81-85%
98%
ee
OH
OH
Scheme 1.13 Osmium-catalyzed dihydroxylation using polyaniline-supported OsO
4
/MTO.
1.3 Supported Osmium Catalyst
j
17
N
H
N
MeO
O
NN
O
N
OMe
N
H
+
(QN)
2
PHAL
mol%OsO0.1
4
(QN)mol%2.5
2
PHAL
NMOeq1.1
acetone/H
2
°C20O/sucrose,
N
H
N
MeO
O
NN
O
N
OMe
N
H
HO HO
OH OH
PhaseSugar
Os
O
O
O
O
OH
OH
PhaseEther
Scheme 1.14 An osmium-(QN)
2
PHAL-sugar recyclable dihydroxylation system.
18
j
1 Recent Developments in Metal-catalyzed Dihydroxylation of Alkenes

including cyclic and acyclic, terminal, mono-, di-, tri-, and tetrasubstituted ones, gave
the corresponding diols in high yields [39]. Enantioselectivities up to 95% ee were
achieved with this type of microencapsulated OsO
4
when (DHQD)
2
PHAL was
introduced as a chiral ligand. However, this system requires slow addition of the
alkene. After running a 100 mmol-scale experiment, more than 95% of the ABS-MC
OsO
4
and the chiral ligand were recovered.
Later, Kobayashi and coworkers reported on a new type of microencapsulated
osmium tetroxide using phenoxyethoxymethyl-polystyrene as support [40]. With
this catalyst, asymmetric dihydroxylation of alkenes has been successfully per-
formed using (DHQD)
2
PHAL as a chiral ligand and K
3
[Fe(CN)
6
] as a cooxidant in
H
2
O/acetone (Scheme 1.16). This dihydroxylation does not require slow addition of
the alkene, and the catalyst can be recovered quantitatively by simple filtration and
reused without loss of activity. With a divinylbenzene-cross-linked polystyrene
microencapsulated OsO
4
and a nonionic phase transfer catalyst (Triton
Ò
X-405),
this system can be run in an aqueous system [41].
A polyurea microencapsulated OsO
4
, so-called Os EnCatÔ, is now commercially
available. Ley et al. demonstrated that this type of microencapsulated OsO
4
catalyzed
dihydroxylation of various alkenes using NMO as the oxidant [42]. Five times
recycling was achieved with good yield (74–88%). With NaIO
4
as the oxidant,
oxidation of alkenes give the corresponding oxidative cleavage products of aldehydes
and ketones.
Microencapsulated OsO
4
can function as a reservoir and an efficient scavenger of
homogeneous catalytic species [42, 43]. Hence, it gives results comparable to those
achieved by its homogeneous analog.
1.3.3
Supports Bearing Alkenes
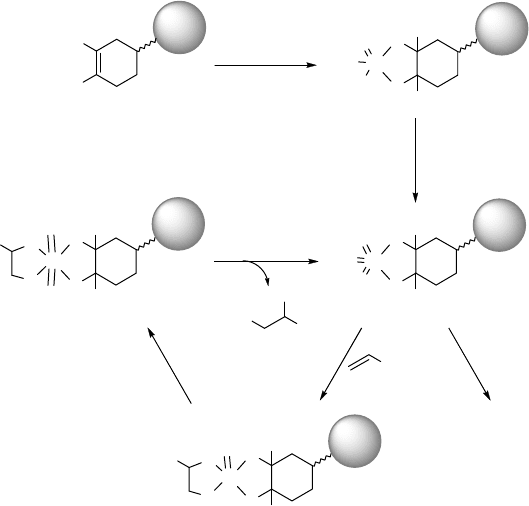
Jacobs and coworkers published a completely different strategy to immobilize
osmium catalysts. Their approach is based on two details of the mechanism of
OsOMC
4
mol%)(5
yield84%
H
2
O-acetone-CH
3
(1/1/1)CN
NMO
h12rt,
OH
OH
Scheme 1.15 Dihydroxylation of cyclohexene using microencapsulated osmium tetroxide
(MC OsO
4
).
OsOPEM-MC
4
mol%)(5
(DHQD)
2
mol%)(5PHAL
H
2
(1/1)O-acetone
K
3
[Fe(CN)
6
K],
2
CO
3
30°C,
R
2
R
1
R
3
R
4
HO OH
R
2
R
4
R
1
R
3
Scheme 1.16 Asymmetric dihydroxylation of alkenes using PEM-MC OsO
4
.
1.3 Supported Osmium Catalyst
j
19
the cis-dihydroxylation: (1) Tetrasubstituted alkenes react with OsO
4
smoothly to
form osmate(VI) esters which are not hydrolyzed under mild conditions, and (2)
an Os(VI) monodiolate complex can be reoxidized to a cis-dioxo Os(VIII) species
without release of the diol. These two properties make it possible to immobilize a
catalytically active osmium compound by the addition of OsO
4
to a tetrasubstituted
alkene which is covalently linked to a silica support. Hence, subsequent addition of a
second alkene results in an Os bisdiolate complex. The tetrasubstituted diolate ester
which is formed at one side of the Os atom is stable, and keeps the catalyst fixed on the
support material. The catalytic reaction can take place at the free coordination sites of
Os (Scheme 1.17) [44].
The dihydroxylation of mono- and disubstituted aliphatic alkenes and cyclic
alkenes is successfully performed using this heterogeneous catalyst and NMO as
cooxidant. The amount of osmium needed is 0.25 mol% Os with respect to the
alkene. This system gives excellent chemoselectivity, being equal to that in the
homogeneous reaction with NMO. However, a somewhat longer reaction time is
necessary. The development of an asymmetric variant of this process by addition of
the typical chiral alkaloid ligands of the asymmetric dihydroxylation would be
difficult, since the reactions performed with these heterogeneous catalysts take
H
3
C
H
3
C
O
O
CH
3
CH
3
Os
O
HO
HO
O
O
CH
3
CH
3
Os
O
O
O
hydrolysisno
O
O
CH
3
CH
3
Os
O
O
O
R
O
O
CH
3
CH
3
Os
O
O
R
O
O
R
OH
HO
OsO
4
[O]
[O]
H
2
O
SiO
2
SiO
2
SiO
2
SiO
2
SiO
2
R
H
2
O
Scheme 1.17 Immobilization of Os in a tertiary diolate complex, and proposed catalytic cycle for
cis-dihydroxylation.
20
j
1 Recent Developments in Metal-catalyzed Dihydroxylation of Alkenes
place in the so-called second cycle. With cinchona alkaloid ligands, high ees are only
achieved in dihydroxylations occurring in the first cycle. However, recent findings by
the groups of Sharpless and Adolfsson show that even second-cycle dihydroxylations
may give substantial ees [45]. Although this process needs to be optimized, further
development of the concept of an enantioselective second-cycle process offers the
prospect of a future heterogeneous asymmetric catalyst.
1.3.4
Immobilization by Ionic Interaction
Choudary and his group reported in 2001 the design of an ion-exchange technique
for the development of recoverable and reusable osmium catalysts immobilized on
layered double hydroxides (LDH), modified silica, and organic resin for asymmetric
dihydroxylation [46]. An activity profile of the dihydroxylation of trans-stilbene with
various exchanger-OsO
4
catalysts revealed that LDH-OsO
4
displays the highest
activity, and the heterogenized catalysts in general have higher reactivity than
K
2
[OsO
2
(OH)
4
]. When trans-stilbene was added to a mixture of LDH-OsO
4
, chiral
ligand (DHQD)
2
PHAL (1 mol% each), and NMO in H
2
O-
t
BuOH, the desired diol
was obtained in 96% yield with 99% ee. Similarly, excellent ees were obtained with
resin-OsO
4
and SiO
2
-OsO
4
in the same reaction. All of the prepared catalysts can be
recovered quantitatively by simple filtration and reused for five cycles with consistent
activity. With this procedure, various alkenes ranging from mono- to trisubstituted,
activated to simple, were transformed into their diols. In most cases, the desired diols
are formed in higher yields, albeit with ees similar to those reported in homogeneous
systems. Slow addition of the alkene to the reaction mixture is sure to achieve higher
ee. This LDH-OsO
4
system presented by Choudary and coworkers is superior in
terms of activity, enantioselectivity, and scope of the reaction to that of Kobayashi.
Although the LDH-OsO
4
shows excellent activity with NMO, it is deactivated when
K
3
[Fe(CN)
6
] or molecular oxygen is used as co-oxidant [47]. This deactivation is
attributed to the displacement of OsO
4
2
by the competing anions, which include
ferricyanide, ferrocyanide, and phosphate ions (from the aqueous buffer solution).
To solve this problem, resin-OsO
4
and SiO
2
-OsO
4
were designed and prepared by the
ion exchange process on the quaternary ammonium-anchored resin and silica,
respectively, as these ion exchangers are expected to prefer bivalent anions over
trivalent anions. These new heterogeneous catalysts show consistent performance
in the dihydroxylation of a-methylstyrene for a number of recycles using NMO,
K
3
[Fe(CN)
6
]orO
2
as reoxidant. The resin-OsO
4
catalyst, however, displays higher
activity than that of the SiO
2
-OsO
4
catalyst. In the presence of Sharpless ligands,
various alkenes can be oxidized enantioselectively using these heterogeneous
systems, and very good ees can be obtained with any of the three cooxidants.
Equimolar ratios of ligand to osmium are sufficient for achieving excellent ees.
This is in contrast to the homogeneous reaction, wherein a 2–3 molar excess of the
expensive chiral ligand to osmium is usually employed. These studies indicate that
the binding ability of these heterogeneous osmium catalysts with the chiral ligand is
greater than that of the homogeneous analog.
1.3 Supported Osmium Catalyst
j
21
Incidentally, this is the first reported heterogeneous osmium-catalyst-mediated
AD reaction of alkenes using molecular oxygen as the cooxidant. Under identical
conditions, the turnover numbers of the heterogeneous catalyst are similar to those
of the homogeneous system.
Furthermore, Choudary and coworkers presented a procedure for the application
of a heterogeneous catalytic system for the AD reaction in combination with
hydrogen peroxide as cooxidant [48]. Here, a triple catalytic system composed of
NMM and two heterogeneous catalysts was designed. Titanium silicalite acts as
electron transfer mediator to perform oxidation of NMM, being used in catalytic
amounts with hydrogen peroxide to provide in situ NMO continuously for AD of
alkenes, which is catalyzed by another heterogeneous catalyst, namely silica gel-
supported cinchona alkaloid [SGS-(DHQD)
2
PHAL]-OsO
4
. Good yields were ob-
served for various alkenes. Again, very good ees are achieved with an equimolar
ratio of ligand to osmium, but slow addition of alkene and H
2
O
2
is advisable.
Unfortunately, recovery and reuse of the [SGS-(DHQD)
2
PHAL]-OsO
4
/TS-1 revealed
that about 30% of the osmium became leached out during the reaction. This amount
has to be replenished in each additional run.
1.4
Ionic Liquid
Recently, ionic liquids have become popular new solvents in organic synthesis [[49,
50]]. They can dissolve a wide range of organometallic compounds and are
miscible with organic compounds. They are highly polar but noncoordinating.
In general, ionic liquids exhibit excellent chemical and thermal stability with e ase
of reuse. It is possible to vary their miscibility with water and organic solvents
simply by changing the counter anion. Their essentially negligible vapor pressure
gives them an advantage over volatile organic solvents from the viewpoint of green
chemistry.
In 2002, a process for alkene dihydroxylation by recoverable and reusable OsO
4
in
ionic liquids was published for the first time [51]. Yanada and coworkers described the
immobilization of OsO
4
in 1-ethyl-3-methylimidazolium tetrafluoroborate [47a].
They chose 1,1-diphenylethylene as a model compound and found that the use of
5 mol% OsO
4
in [emim]BF
4
, 1.2 equiv. of NMOH
2
O, and room temperature were
the best reaction conditions for a good yield. After 18 h, a 100% yield of the cor-
responding diol was obtained. OsO
4
-catalyzed reactions with other co-oxidants such
as hydrogen peroxide, sodium percarbonate, and tert-butyl hydroperoxide gave poor
results. With anhydrous NMO the yield of diol was only 6%. After the reaction, the
1,2-diol can be extracted with ethyl acetate, and the ionic liquid containing the catalyst
can be reused for further catalytic oxidation reaction. It was shown that even in the
fifth run the obtained yield did not change. This new method using immobilized
OsO
4
in an ionic liquid was applied to several substrates, including mono-, di-, and
trisubstituted aliphatic alkenes, as well as to aromatic alkenes. In all cases, the desired
diols were obtained in high yields.
22
j
1 Recent Developments in Metal-catalyzed Dihydroxylation of Alkenes