Arora R. (ed.) Medicinal Plant Biotechnology
Подождите немного. Документ загружается.

14 Plant cell cultures for production of medicinal compounds
decades. However, only a few compounds have reached the commercial production scale,
including shikonin and paclitaxel.
The first recombinant protein, human serum albumin, produced in plants was reported
nearly 20 years ago (Sijmons et al., 1990). Since then, a number of recombinant proteins
have been produced in plant cell cultures. The first technical plant-produced recombinant
proteins to be marketed were avidin, trypsin and C-glucuronidase. Proof-of-concept has
also been established for the plant-based production of many therapeutic proteins including
antibodies, blood products, cytokines, growth factors, hormones, recombinant enzymes and
vaccines. Currently, several plant-produced pharmaceuticals are in clinical trials and
approaching commercial release within the next few years, insulin and glucocerebrosidase
being perhaps the most advanced examples.
Selection of the Production System
Choice of the production system is the first and the most important decision to be made and
highly depends on the compound to be produced. Taking into consideration the long and
expensive path from the initial pharmaceutical/medicinal invention to production on the
market, through clinical stages and regulatory approval, the decision concerning the
manufacturing system must be made at a very early stage (Fig. 2.1). The manufacturing
system carries a long-lasting impact since it affects the viability of the whole chain. The
chosen manufacturing process cannot be changed later without substantial economical loss.
Different production hosts offer different pros and cons, and these factors must be
evaluated against the properties of the final product and against total production costs.
Depending on whether the final product is e.g. a plant-based small molecule or a
therapeutic protein, the method chosen has different requirements. Commercial production
of plant natural compounds using cell and tissue culture systems is calculated to become
viable when a product has high value, with a price exceeding $500–1000/kg (Sajc et al.,
2000). Figure 2.2 presents the advantages displayed by various production hosts to be
considered when choosing a system for production of biomedical compounds.
Various plant secondary metabolites are used as medicinal compounds, due to their
physiological actions. Thus, many efforts have been made to produce them in plant cell
cultures under controlled environment in bioreactors, since these compounds often
accumulate in very low amounts (less than 1% dry weight) in whole plants. Moreover, their
production and accumulation is commonly organ-specific or they are produced only in
specific developmental stages. Many plant-derived pharmaceutical compounds are family-
or even species specific, as for example alkaloids and terpenoids, resulting in highly
versatile designing of production platforms for each species and compound. On the other
hand, the applications of recombinant protein production have mainly been performed
using tobacco plants as expression hosts. However, along the development of plant cell
culture systems, tobacco BY-2 (Nagata et al., 2004) and NT-1 (Nagata and Kumagai,
1999) cell cultures have served as production hosts in a great majority of cases.
In addition to plant cell cultures, alternative production systems for both secondary
metabolites and recombinant proteins are harvest and extraction of the desired compounds
from whole plants, or the use of microbial cells as production hosts.
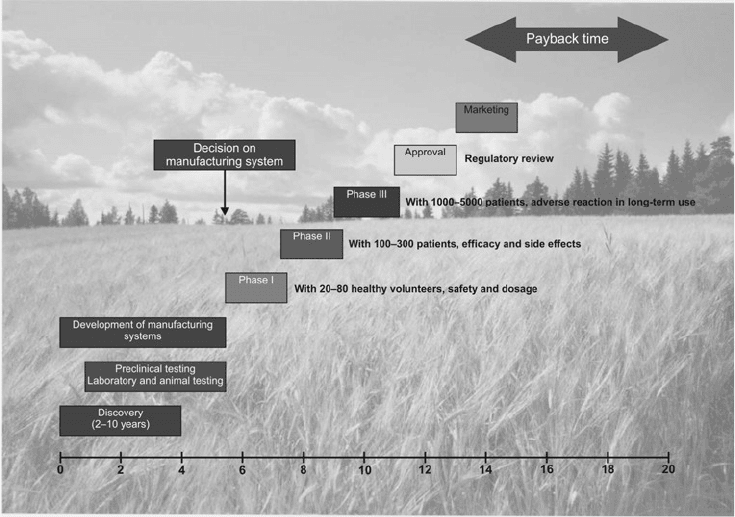
Plant cell cultures for production of medicinal compounds 15
Fig. 2.1. Development of pharmaceuticals from discovery to market launching.
Animal cells have hitherto been the most applied host for recombinant pharmaceutical
protein production. On the other hand, secondary metabolites can also be synthesized
chemically or via semisynthesis. Each method offers distinctive advantages and
disadvantages, depending on the compound of interest, and this will be further discussed in
the following sections.
Production in microbial hosts
If possible, the transfer of biochemical plant pathways to microbial cells offers an
attractive alternative as an environmetally friendly process. Due to their smaller genome
size, the degree of complexity in microorganisms is significantly lower than in plant cells,
and this has generated considerable interest in their exploitation for the synthesis of high-
value plant metabolites. Microorganisms have fewer intracellular organelles compared to
plant cells, and thus metabolite and enzyme compartmentalization can be negligible
(Leonard et al., 2009). In addition, the exploitation of microbial cells offers the possibility
to use inexpensive carbon sources in production processes, allowing the use of simple,
synthetic media. When it comes to large-scale cultivation, microbial cells are less sensitive
to shear stress than plant cells, allowing a wider selection of bioreactors. Moreover, the
shorter doubling time of microbial cells offers faster production rates.
Microbial hosts also have benefits in downstream processing, since typically products
are secreted out of the cells to the surrounding medium, whereas secondary metabolites
produced by plant cells are generally accumulated intracellularly. However, a bottleneck in
the exploitation of microbial cells in the production of plant-based pharmaceuticals is lack

16 Plant cell cultures for production of medicinal compounds
of knowledge of the biosynthetic enzymes and intermediates of the target compounds. For
example, reconstruction of the plant biosynthetic pathway of morphine in microbes,
starting from tyrosine, would require the functional expression of more than 17 enzymes
(Leonard et al., 2009).
Fig. 2.2. Production of biomedical compounds – advantages offered by different production
hosts.
In addition, various enzymatic steps in plant secondary metabolite pathways involve
cytochrome P450, for example the biosynthesis of terpenoids, and the expression of redox-
partners for P450 enzymes in microbes has hitherto been rather challenging (Khosla and
Keasling, 2003). Furthermore, the lack of intracellular S-adenosyl-L-methionine, required
for various methylation reactions typical for alkaloid biosynthesis, might become a limiting
factor in microbial production systems. Efficient production of a plant-derived high-value
drug has been achieved with artemisinin, which is used for the treatment of malaria. Its
worldwide demand has led to severe shortages of natural artemisinin sources. Artemisinin
has not yet been successfully produced in plant cell or tissue cultures with economical
yields. The technology for production of artemisinin and its derivatives in a microbial
system combined with chemical synthesis strategies has been established by expressing
bacterial, yeast and plant genes in Escherichia coli (Chang et al., 2007; Hale et al., 2007)
and yeast (Ro et al., 2006). To illustrate the importance of this kind of technology
development, the Bill and Melinda Gates Foundation granted $42.6 million funds in 2004
to this synthetic biology approach, aiming to get semi-synthetic artemisinin into poor
countries in Asia and Africa by 2012.
Plant cell cultures for production of medicinal compounds 17
Due to its extensive genetic and physiological characterization, short generation time
and established cultivation know-how, E. coli is the most widely applied prokaryotic host
for recombinant protein production. Various pharmaceuticals are being commercially
produced with E. coli, including somatotropines, insulin and interferon gamma (Schmidt,
2004). However, when it comes to recombinant protein production, being prokaryotes,
bacterial cells lack the machinery of plant and animal cells to carry out many desired post-
translational modifications, which limits their use for production of a number of medicinal
recombinant proteins. In addition, the secretion system in E. coli is still not efficient
enough to be exploited in the production of many drugs with high volume demands. Like
E. coli, yeasts can be cultivated rapidly and easily and moreover, their secretion capacity is
considerably higher. Besides Saccharomyces cerevisiae, which is the best characterized
and most extensively used yeast for recombinant protein production, applications using
Pichia as production host have increased rapidly during the past two decades, with more
than 400 proteins reported in 2002 (Lin Cereghino et al., 2002). Examples of commercial
pharmaceutical proteins produced by S. cerevisiae include hepatitis B vaccine and platelet-
derived growth factor (Schmidt, 2004).
Production in animal cells
Animal cell cultures possess the highest similarity to human cells with respect to their
ability to carry out post-translational modifications. Today, approximately 60–70% of all
pharmaceutical recombinant proteins are produced by mammalian cells (Wurm, 2004). The
most frequently applied production host is immortalized Chinese hamster ovary (CHO)
cells. Other cell lines, such as mouse myeloma (NS0), baby hamster kidney (BHK), human
embryo kidney (HEK-293) and human retinal cells have received regulatory approval for
recombinant protein production. Several advances in expression strategies used in
mammalian systems have been made, including improvements in vector constructions and
selectable marker systems, as well as in gene targeting and high-throughput screening
(Andersen and Krummen, 2002). Controllable expression using e.g. tetracycline- or
streptogramin-based gene regulation has been demonstrated to be a useful tool for
multicomponent control strategies and especially for the expression of products which
possess cytotoxic properties (Fussenegger et al., 2000). In addition, recent advances in
glycosylation control (Weikert et al., 1999) and reduced lactate accumulation strategies
(Chen et al., 2001) have led to improved process performance and product quality.
However, the main drawbacks with mammalian production systems are still the relatively
expensive media used in culturing, and overall high production costs. Mammalian cells are
used for commercial production of e.g. blood coagulation factors, erythropoietin and
follitropin (Schmidt, 2004). In addition to mammalian cells, insect cells such as
Trichoplusia have been efficiently used for production of human collagenase IV. Due to
their higher stress resistance and easier handling, insect systems might even be more
desirable options for high-throughput protein expression than mammalian cells (Schmidt,
2004).
Chemical synthesis
During the 25-year period 1981
–2006, a total of 30% of the new chemical entities
approved as drugs were synthetic and 23% were made by semisynthesis (Newman and
18 Plant cell cultures for production of medicinal compounds
Cragg, 2007). Plant-based medicinal compounds can be produced by chemical synthesis if
the stereochemistry or chirality of the compound in question does not limit the use of the
method. However, medicinal plant compounds are often structurally complex, which makes
their chemical synthesis difficult or very expensive. In addition, in many cases synthesis
would require the use of harsh solvents, which may ultimately lead to low product yields.
Further, problems often encountered in the synthetic substitution of natural compounds are
the lack of specificity and efficacy compared to natural compounds. Many natural
compounds have important roles as lead molecules which serve as a backbone for synthetic
derivatives, such as production of irinotecan (an anticancer compound) from camptothecin.
Galanthamine, a drug used in the treatment of Alzheimer’s disease, is an alkaloid that was
initially isolated from the snowdrop (Galanthus woronowii Losinsk.) in the early 1950s,
and it has since also been found from other plants in the Amaryllidaceae family (Howes et
al., 2003). However, due to the limited availability of plants producing this compound,
galanthamine is now produced by total synthesis.
Protein-based drugs are used to diagnose, prevent and treat diseases. The traditional
way of manufacturing these proteins was extraction from animal and plant sources. Today,
after the recombinant technology revolution, safe and cost-efficient large-scale
biotechnological production processes have replaced the old systems. Intense research is
continuing to create synthetic proteins with improved functions and stabilities. The long
term goal is to develop new tailor-made protein therapeutics that could be administered
orally. For this purpose proteins composed of C-amino acids are considered to be one
possibility (Petersson and Schepartz, 2008).
Production in plant cell cultures
Use of plant cell cultures offers an attractive alternative for the production of plant-based
pharmaceuticals. Compared to field-grown plants, cell cultures can be cultivated in a
controlled and contained environment. Production in cell cultures also offers possibilities
for production optimization, independently of climatic or environmental effects (Fig. 2.3).
Problems often associated with extracts from whole plants include lack of reproducibility
in the bioactivities and variation in the biochemical profiles depending on the cultivation
time and location. In addition, valuable compounds in aerial parts of the plants are often
present in very low amounts and their detection can be even more difficult due to the
presence of pigments and polyphenols, which in many cases may interfere in the drug
screens (Poulev et al., 2003).
In many cases the production levels of secondary metabolites in undifferentiated cell
cultures are very low and often secondary metabolism is strongly linked to cellular
differentiation. Moreover, the stability of the production rates in undifferentiated cell
cultures is poor. This is partly related to the somaclonal variation and selection of cells
during long subculturing. As an example, production of the terpenoid indole alkaloids
vincristine and vinblastine has not hitherto been successful in undifferentiated cells and it is
recognized that final dimerization of vindoline and catharantine requires cellular
differentiation (Samanani and Facchini, 2006).

Plant cell cultures for production of medicinal compounds 19
Fig. 2.3. Production of medicinal compounds by plant cell cultures and whole plants.
Hairy root cultures, generated as a result of Agrobacterium rhizogenes infection, have
proved to be good alternatives as production hosts, producing the same compounds and
even in higher quantities than the mother plants (Sevón and Oksman-Caldentey, 2002). In
addition, hairy roots gain biomass rather quickly and have simple cultivation medium
requirements, being able to grow without phytohormones. They commonly display higher
genetic stability and more stable metabolite production than that of undifferentiated cell
cultures. Hundreds of plant species have been transformed with A. rhizogenes for induction
of hairy roots. The majority of cases has involved the use of alkaloid-producing species
(Oksman-Caldentey, 2007).
Medicinal compounds produced by plant cell cultures in commercial or semi-
commercial scale are presented in Table 2.1. In addition, even though not commercially
produced, examples of cell culture systems in which higher production has been reported
than in intact plants include diosgenin by Dioscorea (Sahai and Knuth, 1985) and
ubiquinone-10 by Nicotiana tabacum (Fontanel and Tabata, 1987).
Compared to microorganisms, plant cells are larger in size and have a rigid cell wall,
which makes them more sensitive to shear stress during bioreactor cultivation and mixing.
Another major challenge in exploitation of plant cell cultures as production hosts for
secondary metabolites is the high variability often encountered in production rates. Cells
often tend to form aggregates composed of several individual cells. The individual cells
within aggregates are exposed to differential microenvironmental conditions, which
increases the risks for labile production (Roberts, 2007).
In addition, the slow multiplication rate of plant cells limits the rate of product
accumulation. Methods to increase the yield of plant secondary metabolites and
recombinant proteins obtained by cell culture systems include screening and selection of
high-producing cell lines, precursor feeding, optimization of the nutrient medium and
20 Plant cell cultures for production of medicinal compounds
cultivation parameters, elicitation and immobilization (Rao and Ravishankar, 2002). As an
example, a two-phase cultivation system, in which cells are first cultivated in the medium
optimal or preferable for cell growth, and then changed into production medium, can be
exploited. This strategy has successfully been exploited in the production of shikonin
(Tabata and Fujita, 1985), anthocyanins (Hirasuna et al., 1991), indole alkaloids (Asada
and Shuler, 1989) and paclitaxel (Tabata, 2004).
Optimization of Production in Plant Cell Cultures
Although plant cell cultures have been extensively studied for their possibilities to produce
medicinal high-value compounds, so far very few applications have reached the stage of
commercial production. The problem most frequently encountered is the low production
rate and yield, and many efforts have been made to increase production levels in plant cell
cultures (Table 2.2). These include optimization of the cultivation medium, especially
levels of sugar, nitrate and phosphate, as well as other culturing parameters such as light,
temperature, hormones and aeration (see e.g. Fujita and Tabata, 1987; Rao and
Ravishankar, 2002). Immobilization of producing cells has been achieved by using calcium
alginate beads, stainless steel and foam particles (Hall et al., 1988). Such platforms are still
rather expensive and require product accumulation in the culture medium, which is often
challenging in plant cell culture systems, since in most cases the products formed in plant
cells are stored inside the cells. Nevertheless, the main advantage in immobilized systems is
that the producing cells can be separated from product allowing continuous or semi-
continuous production. Transient permeabilization of cells has been achieved using e.g.
organic solvents or polysaccharides such as chitosan to enhance the release of intracellular
product. In addition, metabolic engineering can be exploited for active transportation of
products outside the cells, by engineering the specific transporter proteins functioning in
secondary metabolism (Yazaki, 2006).
Secondary metabolite production in plants is often highly inducible, and this can be
exploited by eliciting the cultures with elicitor compounds, such as jasmonates and their
derivatives, salicylic acid, chitosan, heavy metals or with biotic elicitors, such as bacterial
or fungal extracts. Jasmonates, octadecanoid-derived signalling molecules, are capable of
inducing the biosynthesis of a wide range of secondary compounds, such as alkaloids,
terpenoids, flavonoids and anthraquinones (Gundlach et al., 1992). In addition to the
production enhancement, elicitor induction has been extensively used for modern
functional genomics-based strategies to generate differential transcriptional expression of
genes involved in the biochemical pathway of the product (Yamazaki and Saito, 2002;
Goossens et al., 2003; Achnine et al., 2005; Rischer et al., 2006; Saito et al., 2007). These
studies have led to many advances in mapping of plant secondary metabolite pathways,
including biosynthesis of anthocyanins, terpenoids and alkaloids.
Increased product formation has also been achieved by feeding cultures with putative
product precursors or intermediates. As an example, phenylalanine addition caused
increased production of rosmarinic acid in cell cultures of Salvia officinalis and Coleus
blumei (Ellis and Towers, 1970; Ibrahim, 1987), and of paclitaxel in Taxus cuspidata
cultures (Fett-Neto et al., 1994).

Plant cell cultures for production of medicinal compounds 21
However, feeding putative precursors, e.g. amino acids or even more proximate
precursors to cultures does not always result in elevated levels of the end products
(Lockwood and Essa, 1984; Hamill et al., 1990). This may be due to the complex
metabolite regulation and feedback systems in plant cells, or sometimes to the high toxicity
of the administered precursor (Robins et al., 1987; Chintapakorn and Hamill, 2003).
Table 2.1. Commercially or semi-commercially cell culture-produced plant medicinal
compounds (adapted from Eibl and Eibl, 2002, 2008; Basaran and Rodríguez-Cerezo,
2008).
Compound Species Manufacturer Reference
Catharanthine Catharanthus
roseus
Mitsui
Chemicals
(Japan)
Misawa,
1994
Echinacea
polysaccharides
Echinacea
purpurea,
Echinacea
angustifolia
Diversa
(Germany)
Ritterhaus et
al., 1990
Geraniol Geranium sp. Mitsui
Petrochemicals
Ind. (Japan)
Misawa,
1994
Paclitaxel Taxus
brevifolia,
Taxus
chinensis
ESCAgenetics
(USA),
Phyton
Biotech(USA/
Germany),
Nippon Oil
(Japan)
Smith, 1995;
Tabata, 2004
Podophyllotoxin Podophyllum
sp.
Nippon Oil
(Japan)
Misawa,
1994
Protoberberines Coptis
japonica
Mitsui
Petrochemical
Ind. (Japan)
Sato and
Yamada,
1984
Rosmarinic acid Coleus blumei Nattermann
(Germany)
Ulbrich et al.,
1985
Scopolamine Duboisia sp. Sumitomo
Chemical
(Japan)
Misawa,
1994
Secondary
compounds
Shikonin Lithospermum
erythrorhizon
Mitsui
Petrochemicals
Ind. (Japan)
Fujita and
Tabata,
1987; Payne
et al., 1991
Glucocerebrosidase Daucus carota Protalix
Biotherapeutics
(Israel)
Shaaltiel et
al., 2007
Recombinant
proteins
ADDC
1
-mediated
antibodies
Physcomitrella
patens
Greenovation
(Germany)
Nechansky
et al., 2007;
Schuster et
al., 2007
1
Antibody-dependent direct cytotoxicity

22 Plant cell cultures for production of medicinal compounds
The other method exploiting the cell’s own metabolic capacity to convert available
substrates into more valuable end-products is called biotransformation (Hamada et al.,
1998; Giri et al., 2001). Plant cells have an excellent capacity to catalyze stereospecific
reactions, resulting in optically pure compounds. In addition, they can carry out
regiospecific modifications that are not easily achieved by chemical synthesis or by e.g.
microorganisms. As an example, C-methyldigitoxin has been converted into much more
valuable cardiac glycoside C-methyldigoxin by Digitalis lanata cell cultures (Alfermann et
al., 1980), and coniferyl alcohol into podophyllotoxin by Podophyllum hexandrum cell
cultures (Van Uden et al., 1995).
Table 2.2. Strategies to enhance the production of pharmaceuticals in plant cell cultures.
Secondary metabolites Recombinant proteins
Selection of high-producing cell lines Codon optimization of foreign gene
sequences
Growth medium and environment
optimization
Protein fusions to improve stability
Immobilization of cells Protein targeting
Use of elicitors Use of inducible promoters
Precursor feeding Growth medium and environment
optimization
Permeabilization of cells
Use of protein antidegradation agents
Metabolite adsorption Use of protein synthesis stimulants
Metabolic engineering Use of secretion inhibitors (brefeldin A)
Recombinant proteins have been produced by plant cell cultures with levels up to 4% of
total soluble protein, although the productivities can vary greatly (Hellwig et al., 2004).
Whether the product is produced inside the cells or secreted to the culture medium is an
important factor affecting the downstream processing. In some cases, downstream
processing can cause the majority of the overall process expenses. However, unlike in the
case of secondary compounds, for which product secretion in the culture medium is
generally a considerable benefit, secretion may seriously alter the stability of the produced
recombinant protein due to protein degradation in the culture medium (Tsoi and Doran,
2002). Hence, sometimes protein targeting into the endoplastic reticulum may result in
higher levels than in the case of secretion to the medium (Schouten et al., 1996; Streatfield,
2007). On the other hand, recovery of the product from plant cell suspensions is commonly
considered to be easier than from microbial suspensions, due to less interfering proteins in
the supernatant. Certain stabilizing agents including dimethylsulfoxide and polyethylene
glycol or secretion inhibitors such as brefeldin A added to the culture medium may
improve the product recovery (see Hellwig et al., 2004 and references therein).
Plant Metabolic Engineering
The major challenge in efficient production of plant secondary metabolites is the discovery
of the biosynthetic steps leading to the final product formation. Hitherto, only a few
pathways have been well characterized, including flavonoid and terpenoid indole alkaloid
Plant cell cultures for production of medicinal compounds 23
(e.g. vincristine) as well as isoquinoline alkaloid (e.g. berberine, morphine) pathways
(Grothe et al., 2001; Winkel-Shirley, 2001; Samanani and Facchini, 2002; Hashimoto and
Yamada, 2003). These achievements have been reached as a result of very long and
laborious research work. Although metabolic engineering has been very extensively
applied in microbes, the properties of higher eukaryotic cells such as plant cells set unique
challenges, which have complicated the discovery of plant metabolic pathways. In plants,
the metabolic pathways are often long and coordinately regulated by several enzymes.
Furthermore, the enzymes often have high specifities for their substrates, which can be
difficult to obtain, and they have low abundance in plant cells and are sometimes very
labile for use in research purposes. In order to successfully perform metabolic engineering
in plant cells, the regulation of multiple steps in parallel or engineering of the regulatory
genes, such as transcription factors, controlling the complete metabolic pathways is needed.
Metabolic profiling has become an integral part of plant functional genomics (Fiehn et
al., 2000; Oksman-Caldentey and Saito, 2005). However, until recently, almost all plant
metabolomics studies have been applied to primary metabolites. Compared to primary
compounds, profiling of secondary compounds is far more challenging, due to their highly
divergent chemical structures and sensitivities in extraction conditions. The huge variety of
different chemical structures, possessing a range of physical and chemical properties, sets
great challenges for analytical tools when profiling multiple metabolites in parallel
(Oksman-Caldentey et al., 2004). Currently, no single analytical technique provides the
ability to profile the complete metabolome and this obstacle has been addressed by using
selective extraction and combination of analytical platforms. The key for understanding
pathway regulation is to define intermediates and to measure flux through the pathway. By
fluxomics approaches integrated with transcriptomic data, a novel means for mapping
pathways at the systems level from gene to metabolite can be created.
In the case of medicinal plants, the first breakthrough example of metabolic engineering
was achieved by Yun et al. (1992). They cloned the gene H6H (hyoscyamine-6C-
hydroxylase) from Hyoscyamus niger and transferred it into Atropa belladonna, a well-
known hyoscyamine-producing species. As a result, the majority of the hyoscyamine was
converted into scopolamine. Later, this finding resulted in the engineering of this final step
in the tropane alkaloid pathway in various other Solanaceae species (Jouhikainen et al.,
1999; Zhang et al., 2004; Oksman-Caldentey, 2007). Recent development of functional
genomics tools as well as the sequencing of the full genome of model plants such as
Arabidopsis and Medigaco, and crops such as rice, maize, soybean and many others in the
near future, can ameliorate the pathway elucidation by a systems biology approach.
However, for many medicinal plants, there is only very limited or no existing data of ESTs
or entire genome sequences. For these purposes, differential display methods, such as
cDNA-AFLP (Yamazaki and Saito, 2002; Goossens et al., 2003; Rischer et al., 2006) have
proved to be excellent tools. The advantages for genome-wide expression analysis methods
such as cDNA-AFLP include the possibility for quantitative transcript profiling and,
moreover, it is applicable to any organism without the need for prior gene sequence
information. It also allows the discovery of completely novel genes, which is very
important when discovering unknown steps in the biosynthesis. Recent advances in high-
throughput genome sequencing methods, such as pyrosequencing (Ronaghi et al., 1998;
Vera et al., 2008), will allow novel means for gene discovery, giving close to one million
400 bp sequences per run with the possibility to analyse multiple samples in parallel.