Allman E.S., Rhodes J.A. Mathematical Models in Biology: An Introduction
Подождите немного. Документ загружается.

176 Constructing Phylogenetic Trees
.32
.18
.24
.02
.32
.18
.24
.02
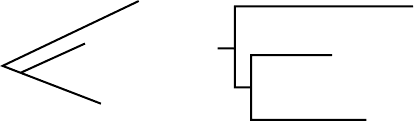
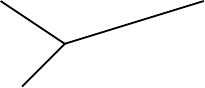
Figure 5.4. Alternate depictions of the same metric tree.
between splittings of the lineage. The longer an edge is, the more the DNA
sequence mutated in the course of the evolution that edge represents.
If, for instance, the Jukes-Cantor model of base substitution adequately
described the evolution of several taxa, then the edge length in a tree relating
them might be the Jukes-Cantor distance between the sequences at the two
ends of the edge. As we saw in Chapter 4, this distance is the average number of
base substitutions per site that occurred in the descent. Included in this are the
mutations obscured by other mutations that the distance formula was designed
to estimate. Because the Jukes-Cantor distance is additive and symmetric,
the total distance between two taxa along a tree should be the Jukes-Cantor
distance between them.
If the molecular clock assumption holds for the evolution of the sequences
being related, then the distances in a tree have a more direct meaning. Recall
that a molecular clock just means that the mutation rate is constant for all
lineages under consideration. If µ denotes the mutation rate, measured in
(base substitutions per site)/year, for instance, and t denotes a time in years,
then the amount of mutation that will occur during this time is
d = µt base substitutions per site.
Thus, a molecular clock means that the amount of mutation along any edge is
proportional to the elapsed time, with the constant of proportionality being the
constant rate of mutation. Under the assumption of a molecular clock, then,
whether we draw edge lengths representing amount of mutation or elapsed
time, we draw exactly the same figure, up to scaling by this constant.
If the molecular clock hypothesis holds for a rooted metric tree, then every
leaf will be located the same total distance from the root. This is because
distances from the root are proportional to the elapsed time since the taxa
began to diverge from the common ancestor. Every taxon has had the same
amount of time to evolve from the root ancestor, so each will have accumulated
the same amount of mutation.
5.1. Phylogenetic Trees 177
Without a molecular clock, the relationship between the amount of mu-
tation along an edge and the amount of time may be complicated. Suppose
that, along one edge of a phylogenetic tree, the mutation rate was quite small,
and along another, the mutation rate was large. Then, even though both edges
might correspond to the same amount of time, considerably more mutation
would occur along one. Without somehow getting additional information
about the rate of mutation – perhaps by comparisons to the fossil record –
we usually do not have ways of determining elapsed times associated to tree
edges.
Metric trees are sometimes drawn in a “square” manner so that it is easier
to compare distance along various evolutionary paths. As an example, the two
trees in Figure 5.4 both represent the same information. In the tree on the left,
the edges have specified lengths, and in the tree on the right, the horizontal
edges have those same lengths. Thus, the vertical edges on the right-hand tree
are read as contributing nothing to the amount of mutation; they serve solely
to separate the various lineages for increased readability.
Problems
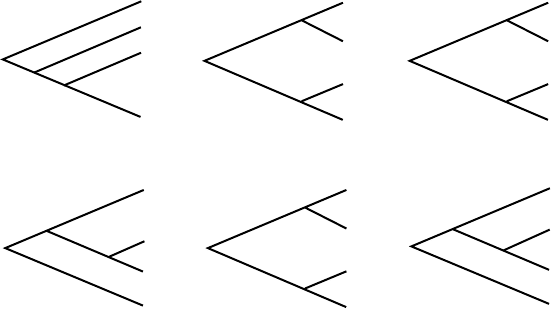
5.1.1. Consider the trees in Figure 5.5.
a. Which of them are the same, as rooted metric trees?
b. Which of them are the same, as unrooted metric trees?
T
1
T
3
T
2
T
5
T
6
.3
.3
.2
.4
.4
.4
T
4
.1
.3
.1
.1
.4
.1
.4
.2
.2
.5
.2
.2
.8
.4
.6
.1
.1
C
B
D
A
A
C
B
D
.3
.5
.2
.3
.4
.4
.2
.5
.3
.2
.4
.4
.2
A
C
D
B
C
A
D
B
A
C
B
D
B
A
C
D
Figure 5.5. Trees for Problem 5.1.1.

178 Constructing Phylogenetic Trees
c. Which of them are the same, as rooted topological trees?
d. Which of them are the same, as unrooted topological trees?
e. For which trees does a molecular clock appear to be operating?
5.1.2. a. Draw the single topologically distinct unrooted bifurcating tree that
could describe the relationship between 3 taxa.
b. Draw the three topologically distinct rooted bifurcating trees that
could describe the relationship between 3 taxa.
5.1.3. a. Draw all 3 topologically distinct unrooted bifurcating trees that
could describe the relationship between 4 taxa.
b. Draw all 15 topologically distinct rooted bifurcating trees that could
describe the relationship between 4 taxa.
5.1.4. For n terminal taxa, the number of unrooted bifurcating trees is
1 · 3 · 5 ···(2n − 5) =
(2n − 5)!
2
n−3
(n − 3)!
.
Make a table of values and graph this function for n ≤ 10.
5.1.5. For n terminal taxa, the number of rooted bifurcating trees is
1 · 3 · 5 ···(2n − 3) =
(2n − 3)!
2
n−2
(n − 2)!
.
Make a table of values and graph this function for n ≤ 10.
5.1.6. In this problem, we will step through the reasoning behind the formulas
for the number of topologically distinct trees, rooted and unrooted.
a. Suppose we already know that an unrooted tree with n terminal
vertices is made up of e edges. Explain why an unrooted tree with
n + 1 terminal vertices will have e + 2 edges. (Hint: Think about
how adding one more terminal vertex to an existing tree affects the
number of edges.)
b. Because an unrooted tree with 2 terminal vertices has 1 edge, explain
from part (a) why an unrooted tree with n terminal vertices will have
1 + 2(n − 2) = 2n − 3 edges.
c. Suppose we know there are m unrooted trees with n terminal ver-
tices. Explain why there will be (2n − 3)m unrooted trees with n + 1
terminal vertices. (Hint: Think about how many different ways you
can add one more terminal vertex to an existing tree.)
d. Because there is only 1 unrooted tree with 2 terminal vertices, ex-
plain from part (c) why there are 1 · 3 · 5 ···(2n − 5) unrooted trees
with n terminal vertices when n > 2.
5.1. Phylogenetic Trees 179
e. Explain why
1 · 3 · 5 ···(2n − 5) =
(2n − 5)!
2
n−3
(n − 3)!
.
f. Why is the number of rooted trees with n terminal vertices the same
as the number of unrooted trees with n + 1 terminal vertices?
g. Conclude that the formulas of Problems 5.1.4 and 5.1.5 are correct.
5.1.7. Because mitochondrial DNA in humans is inherited solely from the
mother, it can be used to construct a tree relating any number of humans
from different ethnic groups, assuming we all descended from a single
first human female. Depending on the clustering pattern of the ethnic
groups, this might give insight into the physical location of this woman
sometimes called Mitochondrial Eve.
In (Cann et al., 1987), a work that first purported to locate Mito-
chondrial Eve in Africa, supporting the “out of Africa” theory of human
origins, a rooted tree was constructed that was claimed to show the rela-
tionships between 147 individuals. How many topologically different
trees would need to be looked at if every possibility was really exam-
ined? (You may need to use Stirling’s formula: n! ∼
√
2πn
n+
1
2
e
−n
.
Here, the symbol “∼” can be interpreted as “is approximately.”)
See (Gibbons, 1992) for the fall-out from the difficulty of consid-
ering so many trees.
5.1.8. The phylogeny of four terminal taxa A, B, C, and D are related ac-
cording to a certain metric tree. The total distances between taxa along
the tree have been found to be as in Table 5.1.
a. Using any approach you wish, determine the correct unrooted tree
relating the taxa, as well as all edge lengths. Explain how you rule
out other topological trees.
b. Can you determine the root from this data? Explain why or why not.
Note: Techniques for this sort of problem are the subject of the next
few sections.
Table 5.1. Distances Between Taxa for
Problem 5.1.8
ABCD
A .6.6.2
B.4.6
C.6

180 Constructing Phylogenetic Trees
5.2. Tree Construction: Distance Methods – Basics
In constructing a phylogenetic tree, the taxa we wish to relate are usually
ones currently living. We have information, such as DNA sequences, from
the terminal taxa and no information from the ones represented by internal
vertices. Indeed, we do not even know which internal vertices should exist,
because we do not yet know the tree topology.
The first class of methods for constructing phylogenetic trees that we will
discuss are distance methods. These attempt to build a tree using information
that we believe describes the total distances between terminal taxa along the
tree.
To see how we might obtain these distances, imagine trying to find the
evolutionary relationship of four species: S1, S2, S3, and S4. Choosing a
particular orthologous stretch of DNA from their genomes, we obtain and
align sequences from each. If the Jukes-Cantor model of base substitution
discussed in Chapter 4 seems appropriate for the data, we then compute Jukes-
Cantor distances between each pair of sequences. These are our estimates of
distances along the tree, which we organize in Table 5.2.
Table 5.2. Distances Between Taxa
S1 S2 S3 S4
S1 .45 .27 .53
S2 .40 .50
S3 .62
Depending on the sequence data, we might instead adopt a different model
of base substitution, leading us to use a different distance formula, such as
the Kimura 2-parameter or the log-det distance. Regardless, the distance we
calculate between sequences is believed to be a measure of the amount of
mutation that has occurred. If these distances were an exact measure of the
amount of mutation that occurred, they would match up with the total distances
between terminal taxa in the metric tree we would to find.
We do not really expect to find a tree that this data fits exactly; after all, the
distances are inferred from sequence data and are not expected to be exactly
correct. Moreover, the method of inferring the distances depended on a model
that involved assumptions that are certainly not met in real organisms. We
hope that however we construct a tree will not be too sensitive to these sorts
of errors in the distances.
UPGMA. The first method we consider is called the average distance
method, or, more formally, the unweighted pair-group method with arithmetic

5.2. Tree Construction: Distance Methods – Basics 181
means (UPGMA). This method produces a rooted tree and assumes a mole-
cular clock. The easiest way to understand the algorithm is by following an
example of its use.
With the data table above, we pick the two closest taxa, S1 and S3. Because
they are .27 apart, we draw Figure 5.6 with each edge .27/2 = .135 long.
.135
.135
S1
S3
Figure 5.6. UPGMA; step 1.
We then combine S1 and S3 into a group, and average the distances of S1
and S3 to each different taxon to get the distance from the group to that taxon.
For example, the distance between S1–S3 and S2 is (.45 + .40)/2 = .425,
and the distance between S1–S3 and S4 is (.53 + .62)/2 = .575. Our table
thus collapses to Table 5.3.
Table 5.3. Distances Between
Groups; UPGMA, Step 1
S1–S3 S2 S4
S1–S3 .425 .575
S2 .50
Now, we simply repeat the process, using the distances in the collapsed
table. Because the closest taxa and/or groups in the new table are S1–S3 and
S2, which are .425 apart, we draw Figure 5.7.
S1
S3
S2
.135
.135
.0775
.2125
Figure 5.7. UPGMA; step 2.
182 Constructing Phylogenetic Trees
The edge to S2 must have length .425/2 = .2125, while the other new
edge must have length (.425/2) − .135 = .0775, because we already have
the edges of length .135 to account for some of the distance between S2 and
the other taxa.
Again combining taxa, we form a group S1–S2–S3, and compute its dis-
tance from S4 by averaging the original distances from S4 to each of S1,
S2, and S3. This gives us (.53 + .5 + .62)/3 = .55. (Note that this is not the
same as averaging the distance from S4 to S1–S3 and to S2.) Because a new
collapsed distance table would have this as its only entry, there is no need to
give it. We draw Figure 5.8, estimating that S4 is .55/2 = .275 from the root.
The final edge has length .0625, since that places the other taxa .275 from the
root as well.
S1
S3
S2
S4
.135
.135
.0775
.0625
.275
.2125
Figure 5.8. UPGMA; step 3.
As we suspected, the tree we have constructed for the data does not exactly
fit the data. The distance on the tree from S3 to S4, for instance, is .55, although
according to the original data, it should be .62. Nonetheless, the tree distances
are at least reasonably close to the distances given by the data.
If we had more taxa to relate, we would have to do more steps to complete
the UPGMA process, but there would be no new ideas involved. At each step,
we join the two closest taxa or groups together, always placing them at equal
distances from a common ancestor. We then collapse the joined taxa into a
group, using averaging to compute a distance from that group to the taxa and
groups still to be joined. The one point to be particularly careful about is
that when the distances between two groups are computed, we must average
all the distances from members of one group to another – if one group has
n members and another has m members, we have to average nm distances.
Each step of the algorithm reduces the size of the distance table by one, so
that after enough steps, all of the taxa are joined into a single tree.
5.2. Tree Construction: Distance Methods – Basics 183
Notice that the molecular clock assumption is implicit in UPGMA. In this
example, when we placed S1 and S3 at the ends of equal length branches, we
assumed that the amount of mutation each underwent from their common an-
cestor was equal. UPGMA always places all the taxa at the same distance from
the root, so that the amount of mutation from the root to any taxon is identical.
Fitch-Margoliash algorithm. This method is a bit more complicated than
UPGMA, but builds on its basic approach. However, it attempts to drop the
molecular clock assumption of UPGMA.
Before giving the algorithm, we need a few mathematical observations.
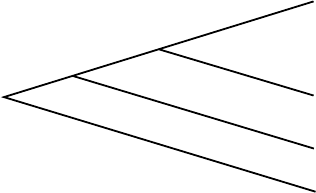
First, if we attempt to put 3 taxa on an unrooted tree, then there is only one
topology that needs to be considered. Furthermore, for 3 taxa, we can assign
lengths to the edges to fit the data exactly. To see this, consider the tree in
Figure 5.9. If we have some distance data d
AB
, d
AC
, and d
BC
, then
x + y = d
AB
,
x + z = d
AC
,
y + z = d
BC
.
These equations can be solved either by writing the system as a matrix equa-
tion and finding an inverse, or by substituting formulas for one variable ob-
tained from one of the equations into the others. Either way leads to the
solution
x = (d
AB
+ d
AC
− d
BC
)/2,
y = (d
AB
+ d
BC
− d
AC
)/2,
z = (d
AC
+ d
BC
− d
AB
)/2.
(5.1)
We will refer to these formulas as the 3-point formulas for fitting taxa to
a tree. Unfortunately, with more than 3 taxa, exactly fitting data to a tree is
usually not possible. The Fitch-Margoliash (cited in tables as FM) algorithm
uses the 3 taxa case, however, to handle more taxa.
Now we explain the operation of the algorithm with an example. We’ll use
the distance data in Table 5.4.
A
B
C
x
y
z
Figure 5.9. The unrooted 3-taxon tree.

184 Constructing Phylogenetic Trees
Table 5.4. Distances Between Taxa
S1 S2 S3 S4 S5
S1 .31 1.01 .75 1.03
S2 1.00 .69 .90
S3 .61 .42
S4 .37
We begin by choosing the closest pair of taxa to join, just as we did with
UPGMA. Looking at our distance table, S1 and S2 are the first pair to join. In
order to join them without placing them at an equal distance from a common
ancestor, we temporarily reduce to the 3-taxa case by combining all other
taxa into a group. For our data, we thus introduce the group S3–S4–S5. We
find the distance from each of S1 and S2 to the group by averaging their
distances to each group member. The distance from S1 to S3–S4–S5 is thus
d(S1, S3–S4–S5) = (1.01 + .75 + 1.03)/3 = .93, whereas the distance from
S2 to S3–S4–S5 is d(S2, S3–S4–S5) = (1.00 + .69 + .90)/3 = .863. This
gives us Table 5.5.
Table 5.5. Distances Between
Groups; FM Algorithm, Step 1a
S1 S2 S3–S4–S5
S1 .31 .93
S2 .863
With only three taxa in this table, we can exactly fit the data to the tree
using the 3-point formulas to get Figure 5.10. The key point here is that the
3-point formulas, unlike UPGMA, can produce unequal distances of taxa
from a common ancestor.
.7415
.1885
.1215
S1
S2
S3-S4-S5
Figure 5.10. FM algorithm; step 1.
We now keep only the edges ending at S1 and S2 in Figure 5.10 and
return to our original data. Remember, the group S3–S4–S5 was only needed
temporarily so we could use the 3-point formulas; we did not intend to join

5.2. Tree Construction: Distance Methods – Basics 185
those taxa together yet. Because we have joined S1 and S2, however, we
combine them into a group for the rest of the algorithm, just as we would
have done with UPGMA. This gives us Table 5.6.
Table 5.6. Distances Between Groups; FM
Algorithm, Step 1b
S1–S2 S3 S4 S5
S1–S2 1.005 .72 .965
S3 .61 .42
S4 .37
We again look for the closest pair (now S4 and S5) and join them in a similar
manner. We combine everything but S4 and S5 into a single temporary group
S1–S2–S3 and compute d(S4, S1–S2–S3) = (.75 + .69 + .61)/3 = .683 and
d(S5, S1–S2–S3) = (1.03 + .90 + .42)/3 = .783. This gives us Table 5.7.
Applying the 3-point formulas to Table 5.7 produces Figure 5.11.
Table 5.7. Distances Between Groups; FM
Algorithm, Step 2a
S1–S2–S3 S4 S5
S1–S2–S3 .683 .783
S4 .37
S4
S5
S1-S2-S3
.548
.135
.235
Figure 5.11. FM algorithm; step 2.
We keep the edges joining S4 and S5 in Figure 5.11, discarding the edge
leading to the temporary group S1–S2–S3. Thus we now have two joined
groups, S1–S2 and S4–S5. To compute a new table containing these two
groups we have found, we average d(S1–S2, S4–S5) = (.75 + 1.03 + .69 +
.90)/4 = .8425 and d(S3, S4–S5) = (.61 + .42)/2 = .515. We have already
computed d(S1–S2, S3) so we produce Table 5.8. At this point, we can fit a