Pecharsky V.K., Zavalij P.Y. Fundamentals of Powder Diffraction and Structural Characterization of Materials
Подождите немного. Документ загружается.

552
Chapter
6
Table
6.34. Coordinates of atoms in the unit cell of Nd5Si as determined from x-ray powder
diffraction data in the space group symmetry P4,2,2
(RF
=
27.1%).
Atom Site
x
Y
z
Nd 1 8(b) 0.3714 0.0057 0.4561
Nd2 8(b) 0.1280 0.9903 0.8779
Nd3a 4(4 0.3103 0.3103 0
Si 1 8(b) 0.4220 0.2004 0.8095
Si2 8(b) 0.1778 0.2070 0.3108
"
The coordinates of Nd3 were modified from 0.8 103, 0.1897,3/4
(Table
6.33) to represent
the triplet in a standard notation, i.e.
x,
x,
0 (see
Table
6.30), by using the following
transformation:
x-
112, 112-y, z-0.75.
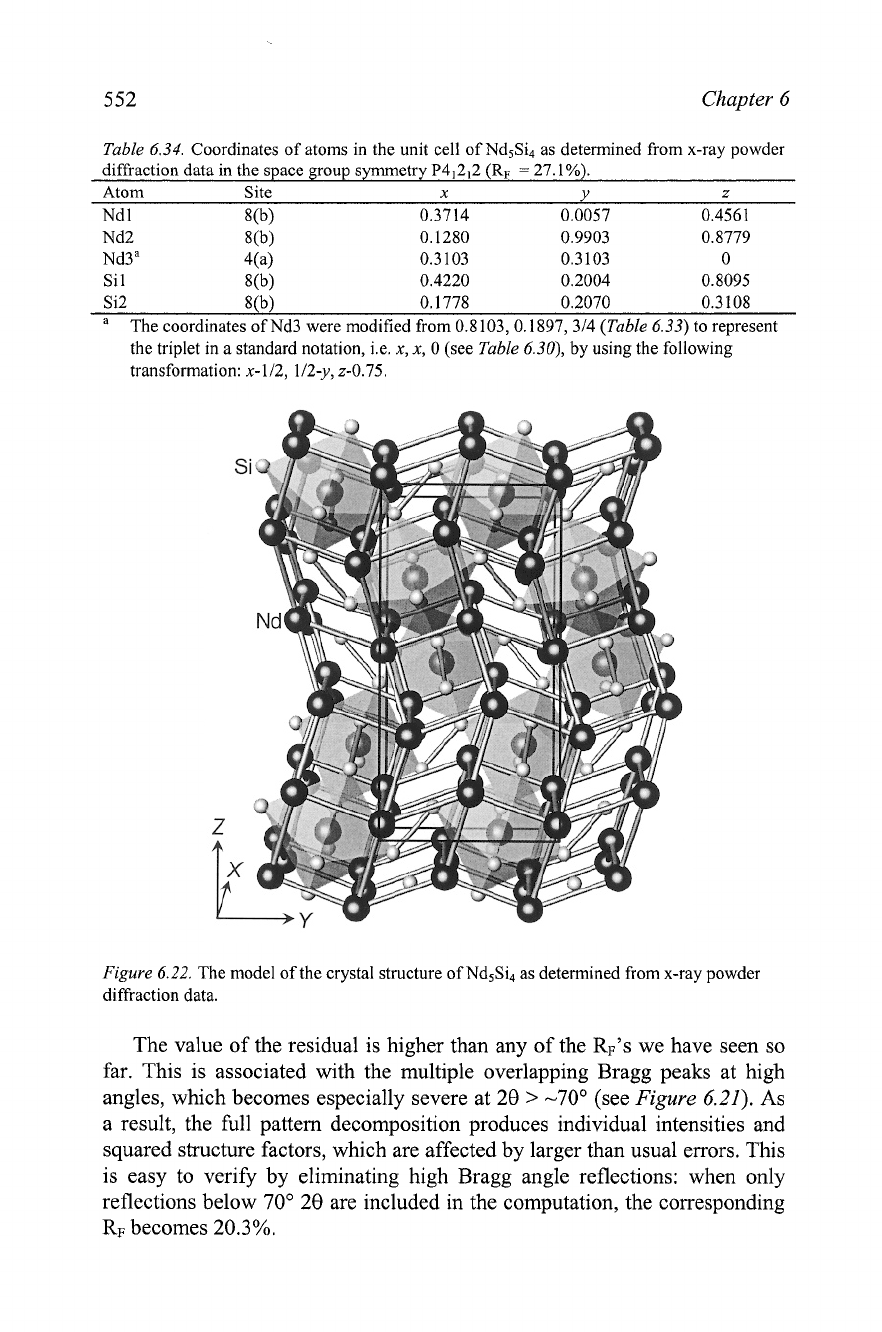
Figure
6.22.
The model of the crystal structure of Nd5Si4 as determined from x-ray powder
difiaction data.
The value of the residual is higher than any of the RF's we have seen so
far. This is associated with the multiple overlapping Bragg peaks at high
angles, which becomes especially severe at 28
>
-70" (see
Figure
6.21).
As
a result, the full pattern decomposition produces individual intensities and
squared structure factors, which are affected by larger than usual errors. This
is easy to verify by eliminating high Bragg angle reflections: when only
reflections below 70" 28 are included in the computation, the corresponding
RF
becomes 20.3%.

Crystal structure solution 553
The Pearson symbol of this crystal structure is tP36 and after consulting
Pearson's Handbook, it is easy to find that the crystal structure of Nd5Si4
belongs to the Zr5Si4-type.' Its model is shown in Figure
6.22.
The solution
of this crystal structure must be completed by Rietveld refinement and it will
be discussed in the next chapter.
6.13
Crystal structure
of
NiMn02(0H)'
This is an example of a quite simple inorganic compound, nickel
manganese oxide hydroxide NiMn02(OH), with relatively good but far from
perfect crystallinity. Due to a peculiar shape of the crystallites, which grow
as well-defined and elongated faceted needles (see inset in Figure
6.23), the
powder exhibits a tendency toward a complex preferred orientation. The
latter is always a complicating factor especially when a structure solution
from first principles should be ~ndertaken.~ This example also illustrates
how to detect and refine hydrogen atoms from powder data (see Chapter 7),
which may be a difficult and often impossible task to accomplish using x-
rays.
Powder diffraction data were collected using a specimen prepared
from a
thoroughly ground powder screened through a 38
pm sieve. First, a fast
experiment was conducted in the range 2
I
29
I
70" using a continuous
scan with a sampling step of 0.03" and a scan rate of 1 deglmin. Positions of
23 individual Bragg reflections at 29
I
60" were determined using a semi-
manual profile fitting. Indexing was performed using TREOR, which
resulted in an orthorhombic base-centered lattice with
24
possible
reflections,
F20
=
135 (0.003,47).
In
order to proceed with the structure solution, a high quality powder
diffraction pattern to 29,,,
=
110" was collected in a weekend experiment
(Figure 6.23, data files
Ch6ExO5-CuKa.xy
and
Ch6ExO5-CuKa.raw
are
located on the CD). The unit cell dimensions were refined using all 75
observed Bragg peaks resulting in a
=
2.8609(1) A,
b
=
14.6482(5) A, c
=
5.2703(2) A,
V
=
220.86(2) A3, and sample displacement of -0.123(6) mm
for a 250
rnrn
goniometer radius.
H.U.
Pfeifer and
K.
Schubert, Crystal structure of Zr5Si4,
Z.
Metallk.
57,
884
(1966).
R. Chen, P.Y. Zavalij, MS. Whittingham,
J.E.
Greedan,
N.P.
Raju, and M. Bieringer, The
hydrothermal synthesis of the new manganese and vanadium oxides,
NiMn03H,
MAV307
and
MAo,75V4010~0.67H20
(MA
=
CH3NH3),
J.
Mater. Chem.
9,
93
(1999).
The
polycrystalline material was prepared by hydrothermally treating a mixture of
Li2C03,
N(CH3)4Mn04,
and
Ni(CH3C00)2
taken in a molar ratio
1:
1.5:
1
at
200•‹C
for
2
days.
When preferred orientation effects are strong, the intensities of Bragg reflections become
biased by systematic errors.
A
correction is nearly impossible before the crystal structure
is solved and the preferred orientation refined using an acceptable model. These errors are
in addition to the errors introduced by deconvolution of the overlapped Bragg peaks.
Chapter
6
10 20 30 40 50 60 70 80 90 100 110
Bragg angle,
20
(deg.)
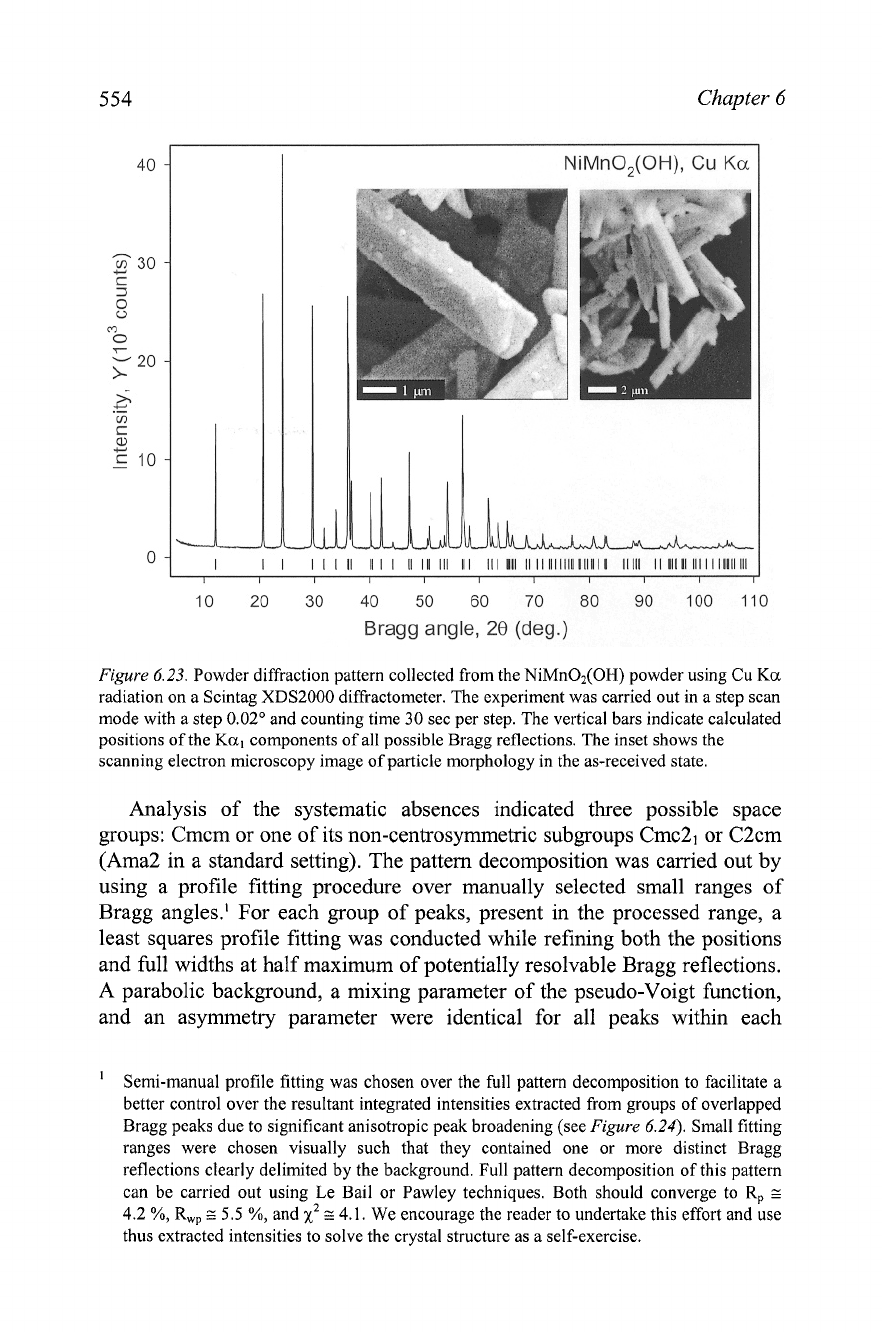
Figure
6.23. Powder diffraction pattern collected from the NiMnO,(OH) powder using Cu
Ka
radiation on a Scintag XDS2000 diffractometer. The experiment was carried out in a step scan
mode with a step
0.02'
and counting time 30 sec per step. The vertical bars indicate calculated
positions of the
Kal
components of all possible Bragg reflections. The inset shows the
scanning electron microscopy image of particle morphology in the as-received state.
Analysis of the systematic absences indicated three possible space
groups: Cmcm or one of its non-centrosymmetric subgroups Cmc2, or C2cm
(Ama2 in a standard setting). The pattern decomposition was carried out by
using a profile fitting procedure over manually selected small ranges of
Bragg angles.' For each group of peaks, present in the processed range, a
least squares profile fitting was conducted while refining both the positions
and full widths at half maximum of potentially resolvable Bragg reflections.
A
parabolic background, a mixing parameter of the pseudo-Voigt function,
and an asymmetry parameter were identical for all peaks within each
'
Semi-manual profile fitting was chosen over the full pattern decomposition to facilitate a
better control over the resultant integrated intensities extracted from groups of overlapped
Bragg peaks due to significant anisotropic peak broadening (see
Figure
6.24). Small fitting
ranges were chosen visually such that they contained one or more distinct Bragg
reflections clearly delimited by the background. Full pattern decomposition of this pattern
can be carried out using Le Bail or Pawley techniques. Both should converge to
Rp
z
4.2
%,
R,.,,
z
5.5
%,
and
x2
E
4.1. We encourage the reader to undertake this effort and use
thus extracted intensities to solve the crystal structure as a self-exercise.
Crystal structure solution
555
selected range. The average
Rp
was
-2.5
%.I
In
order to maintain stability of
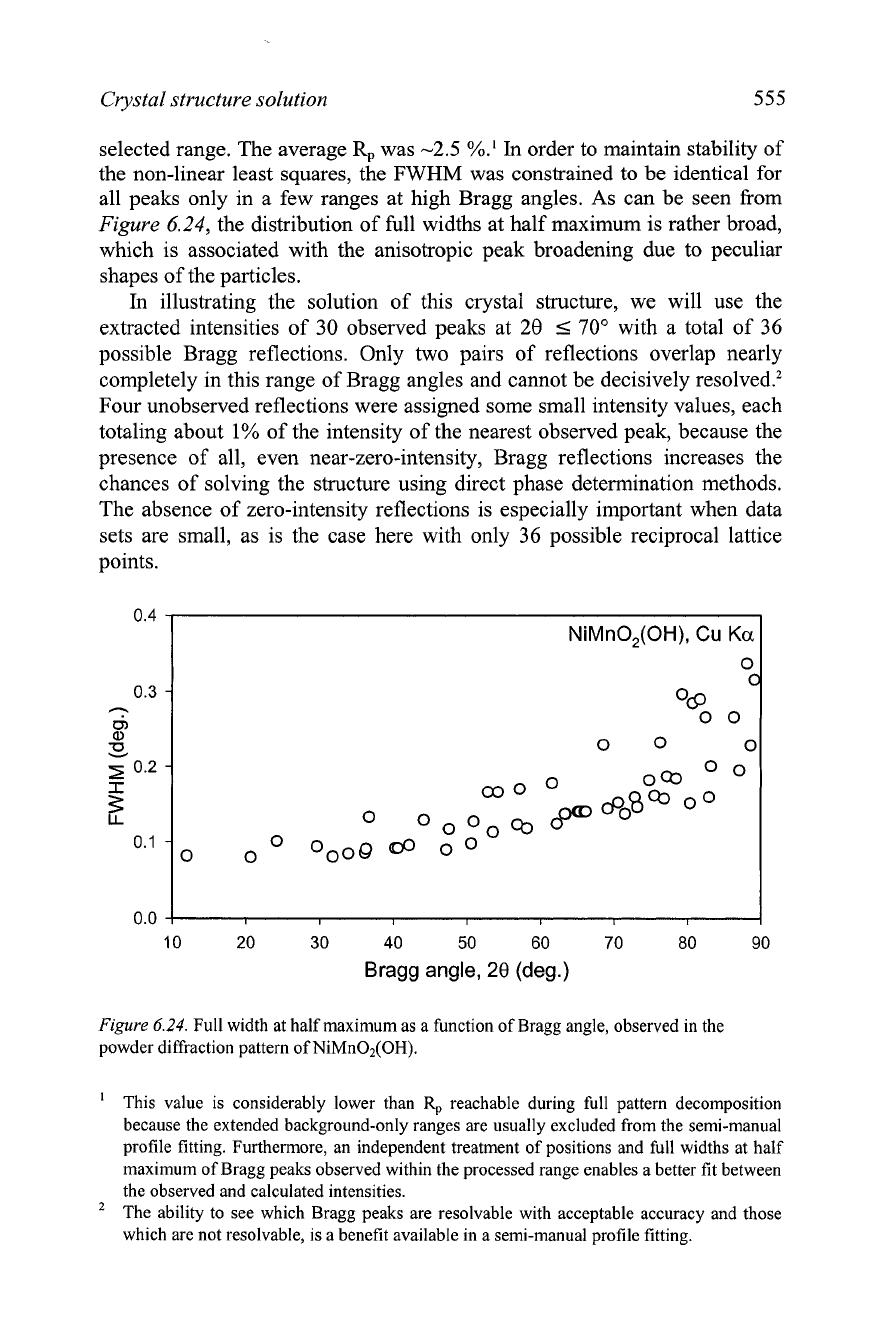
the non-linear least squares, the FWHM was constrained to be identical for
all peaks only in a few ranges at high Bragg angles. As can be seen from
Figure
6.24,
the distribution of full widths at half maximum is rather broad,
which is associated with the anisotropic peak broadening due to peculiar
shapes of the particles.
In
illustrating the solution of this crystal structure, we will use the
extracted intensities of 30 observed peaks at
28
I
70" with a total of 36
possible Bragg reflections. Only two pairs of reflections overlap nearly
completely in this range of Bragg angles and cannot be decisively re~olved.~
Four unobserved reflections were assigned some small intensity values, each
totaling about
1%
of the intensity of the nearest observed peak, because the
presence of all, even near-zero-intensity, Bragg reflections increases the
chances of solving the structure using direct phase determination methods.
The absence of zero-intensity reflections is especially important when data
sets are small, as is the case here with only 36 possible reciprocal lattice
points.
V.V
I
I I
I
I I I I
10 20 30 40 50 60 70 80 90
Bragg angle,
28
(deg.)
Figure
6.24.
Full width at half maximum as a function of Bragg angle, observed in the
powder diffraction pattern of NiMn02(OH).
This value is considerably lower than
R,,
reachable during full pattern decomposition
because the extended background-only ranges are usually excluded from the semi-manual
profile fitting. Furthermore, an independent treatment of positions and full widths at half
maximum of Bragg peaks observed within the processed range enables a better fit between
the observed and calculated intensities.
The ability to see which Bragg peaks are resolvable with acceptable accuracy and those
which are not resolvable, is a benefit available in a semi-manual profile fitting.

556
Chapter
6
The indexed list of the individual structure factors
lFob,l
is found on the
CD in the file Ch6ExOS.hkl and the corresponding crystal data in the file
Ch6ExOS,ins, both are in the
SHELX
format. The intensities of all observed
Bragg reflections may also be employed in the solution of this crystal
structure, even with a greater success. Thus, the corresponding list is found
in the file Ch6ExO5-full.hk1, and we encourage the reader to use it as a
practical self-exercise.
Chemical composition of the crystals was established from microprobe
and thermogravimetric (TGA) analyses. According to microprobe data, the
ratio of Mn to Ni is 1:l. From TGA results, the amount of oxygen was
determined to be 1.5 times that of both metals, thus suggesting a 1:
1
:3
stoichiometry, i.e. NiMn03. The amount of hydrogen cannot be accurately
established due to the complexity of the TGA trace, which has several
different weight losses in both oxygen and nitrogen atmospheres.
The gravimetric density of the crystals was not measured but the content
of the unit cell may be established by using
Eq.
6.5 and the expectation that
the reasonable value of
p
should be between 4 and 5 g/cm3. The estimated
density assuming NiMn03 composition has a reasonable value of 4.86 g/cm3
when
Z
=
4.
The two closest numbers of formula units
(Z
=
3 or 5) are
impossible due to the restrictions imposed by symmetry: in a base-centered
lattice, sites with odd multiplicities are impossible. The next two closest
numbers
(Z
=
2 or 6) result in unrealistically low and high densities,
respectively. Thus, we assume that there are 4 Mn, 4 Ni and 12
0
atoms in
the unit cell.
Considering space group Cmcm first, the multiplicity of the general site
here is
16.
The group also includes several
8-
and 4-fold sites, suggesting
that at least one special position with the multiplicity
4
is occupied by
oxygen. Both Mn and Ni atoms should occupy 4-fold sites. The remaining 8
0
atoms can occupy one position with the multiplicity 8 or two 4-fold sites.
The very short unit cell dimension,
a
E
2.86
8,,
and the presence of mirror
planes perpendicular to it further limits possible locations of atoms in this
unit cell: all atoms should be located in the mirror planes. Thus, only the
following sites 4(a): 0,0,0; 4(b): 0,1/2,0; 4(c): O,y,1/4; and 8(9: O,y,z may be
occupied. If
x
accepts a non-zero value, the distance between symmetrically
related atoms becomes -1.43
8,
or shorter,' which is unrealistic. Another
possibility is to use the non-centrosymmetric groups. The space group C2cm
does not look promising because the absence of the mirror plane
perpendicular to
X,
which is a very short unit cell edge, is highly unlikely as
'
Mirror planes are spaced at
112~.
Thus, every atom with the coordinates
x,y,z
has nearest
symmetrically equivalent atoms at
-x,y,z
and
1-x,y,z.
The pairs of atoms are separated by
2xa
and
(1-&)a,
respectively. The distances are at maximum when
x
=
114,
i.e. the
spacings are
a12
z
1.43
A.

Crystal structure solution
discussed above. The non-centrosymmetric space group Cmc2, looks more
promising as it has a 4-fold special position in the mirror plane perpendicular
to
X:
4(a) with coordinates
Oyz,
where all atoms can be located.
The structure was solved' using SHELXS-90 and partial least squares
refinement using SHELXL-97 programs.' The centrosymmetric space group
symmetry Cmcm was tested first, however, several attempts with varying
parameters produced no acceptable model. It may be difficult to recognize
the incorrect selection of space group symmetry based on a few failures to
find the model, especially when relatively low quality or truncated structure
factor data are employed
(e.g. those extracted from the powder pattern). If a
solution at certain conditions in the selected space group symmetry was not
found, this does not necessarily mean that it does not exist. Often, it may be
tricky to identify a true solution.
Taking into account that the number of formula units per cell
(Z
=
4)
gives a preference to the space group Cmc2,, it was chosen for the next
attempt. At first, the direct phase angles determination using SHELXS-90
was attempted with all default parameters. The program automatically
assigns heavy atoms to the peaks from the E-map, and in this case, the first
three peaks were treated as Mn. Analysis of interatomic distances indicated,
however, that the second strongest peak cannot be a metal and therefore, this
solution was
aband~ned.~
The following step, which is usually recommended when working with
powder data and when the default parameters do not result in an acceptable
solution, is to decrease the minimum normalized structure amplitude (Efi,,)
employed in the generation of phases.
In
general, this reduction decreases
the probability of phase relationships (see Eqs. 2.144, 2.147, and 2.148) but
it increases the number of reflections included in the process.
In
our case,
decreasing E,, from the default 1.2 to Ed,
=
1.1, increases the number of
reflections from 11 to 14. The best solution, shown in
Table
6.35,
contains
the first two peaks that are suitable as metals and the next three can be
suitable as oxygen atoms.4 Peaks beginning from
44
and below are
unacceptable because they are too close to the already assigned peaks.
Ready to use reflection file
Ch6ExOS.hkl
and crystal data file
Ch6ExOS.ins
are found on
the
CD.
Both can be used as input files for SHELXS-90.
G. M. Sheldrick, Phase annealing in SHELXS-90: direct methods for larger structures,
Acta Cryst.
A46,
467 (1990); G. M. Sheldrick, SHELXL-97. University of Gottingen,
Germany, (1997). See the footnote on page
515
on how to obtain the programs.
Generally, a situation like that does not necessarily mean that the model of the crystal
structure cannot be completed using this solution. It may take longer and it may be harder
to make decisions about which peaks should be included, and what atom types should be
assigned to them.
The suitability of peaks as atoms has been judged based on the relative heights of the
peaks on the E-map and from the shortest interatomic distances. The distance Mn2
-
42

558
Chapter
6
Table
6.35.
Maxima localized from the E-map refined for the best solution obtained by
SHELXS-97 using 14 reflections with
Emin
=
I.
1
employing intensity data at
29
5
70'.
Bond distances,
A
Peaka
x
Y
z
Height Comment
~nl~ ~n2~
Mnl 0.0000 0.3018 0.4909 71 1.5
Q5
-0.5000 0.2276 0.3901 60.1
-
a
Mn is indistinguishable from Ni at this stage because the former contains 25 electrons,
while the latter has
28
electrons, i.e. both atoms have similar scattering factors.
This column lists the distances to the indicated atom in
8.
The obtained model was refined by using SHELXL-97 to
RF
=
25
%,
which resulted in the coordinates listed in
Table
6.36. Two oxygen atoms,
02 and 03, converged into locations that are too close to Mnl and therefore,
were eliminated from the model. A difference Fourier map was calculated
using phase angles determined by Mnl, Mn2, and 01 and the first two
strongest maxima are at reasonable distances to both metal atoms.
This is a good place to comment on a nearly blind inclusion of 02 and
03 in the previous step: it was done without the proper analysis of the
geometry of the model,
i.e. bond angles and coordination polyhedra should
be always analyzed in addition to bond distances.' The insertion of
"incorrect" atoms could be more detrimental than the simple removal of
these atoms at a later point. Indeed, this may disable solving a structure as a
whole. Therefore, only those atoms, which are rational from chemical and
physical points of view, should be
includedadded to the model. If there is a
suspicion about a partially built model, a conventional Fourier map should
be computed and used to improve the coordinates of already known atoms
and verify their positioning in the unit cell (e.g. see section 6.10, where the
improper positioning of the
Rh
atom was easily detected from a Fourier map
calculated in the space group 14lmmm).
Two new oxygen atoms improve the geometry of the model and the
following refinement lowers
RF
to 9.5%
(Table
6.37). Overall, this result
gives a great confidence in the correctness of the model. All peaks on the
(1.59
a)
is quite short, but at this point in the structure solution it may be acceptable
considering a small number of reflections included into the computation of both the phases
and E-map.
'
Analysis of bond angles is usually unnecessary in the case of intermetallic structures,
where bonding is predominantly metallic. However, knowing bond angles is critical when
solving structures with principally covalent bonding.
Crystal structure solution
559
subsequent difference Fourier map (only two are listed in
Table
6.37)
are too
close to the already present atoms.
Table
6.36. Results of structure refinement (SHELXL-97) and the difference Fourier peaks
computed using all reflection data below 20
=
70' (RF
=
25
%).
Atom/ Uisd Bond distances,
A
Peak
X
Y
Height Mnla Mn2a 02a 03a
Comment
Mnl 0.0000 0.3021 0.4777
~4 1.0000 0.2146 0.7047 2.8
a
This column lists the distances to the indicated atom in
A.
Deleted.
Table
6.37.
Results of structure refinement (SHELXL-97) and the difference Fourier peaks
computed using all reflection data below 20
=
70' (RF
=
9.5
%).
Atom/
x
Uisd
Bond distances,
A
Com-
Peak
Y
Z
Height Mnla Mn2a Ola 02a ment
Mnl 0.0000
0.3047 0.4759
0.022
Mn2 0.5000
0.5004 0.7235
0.032
01 0.0000
0.4496
0.9497 0.000
2.01,2.16
02 0.5000 0.3920
0.4445 0.029 1.93 1.96,2.17
03 0.5000
0.2177 0.5605
0.007 1.97,2.22
Q1 -0.500 0.4302
0.9673 1.9 1.65 1.46
b
42 0.5000 0.4249
0.6171 1.5 1.24 1.03
"
This column lists the distances to the indicated atom in
A.
Distances are unreasonably short.
Thorough readers who
try
to use the full array of diffraction data in order
to verify the solution of this simple crystal structure will easily find out how
important is the completeness of the data: the whole model may be obtained
from an E-map employing phase angles, generated by using
Efi,
=
1.1
.'
The
refinement of atomic parameters, however, converges to
RF
=
27
%,
which is
much higher than
RF
=
9.5
%
obtained for the data array truncated at
28
=
'
The full array of the individual structure factors is located on the CD in the file
Ch6ExOS-full.hk1.
Identical Bragg reflections have slightly different values of
l~~~,"~~l
when compared to
Ch6ExOS.hkl
because two files were created during two independent
profile-fitting attempts. Readers can use this file, or they can create their own list of the
observed structure factors by using Le Bail or
Pawley full pattern decomposition
approaches. In this example, the anisotropic peak broadening should be refined to achieve
good convergence between the observed and calculated powder diffraction profiles.

560
Chapter
6
70". This increase in RF is associated with the weaker and broader peaks and
with a substantial overlap at high Bragg angles, which disable accurate
determination of the individual integrated intensities.
A
similar situation was
observed in the intermetallic Nd5Si4, see section 6.12.
It is critical to realize that low RF alone, cannot be taken as a sufficient
evidence for the rationality of the structural model.' Far more important
measures of whether the model is correct, are its geometry (bond distances,
angles, coordination polyhedra) and electroneutrality of the crystal.
In
this
example, the structure consists of square pyramids for Mnl and octahedra
for Mn2, which share oxygen atoms and form a three-dimensional
framework as shown in
Figure
6.25.
Furthermore, this figure clearly
illustrates that there are no mirror planes perpendicular to
Z
and confirms the
correctness of the space group Cmc2,. The resulting model appears
reasonable and complete except Mn and Ni atoms must be distinguished, and
locations of hydrogen atoms should be established. The latter may require a
different type of experimental data, while the former should be doable
during Rietveld refinement of the structural parameters using the existing x-
ray diffraction data, as will be discussed in Chapter 7, section 7.7.
Figure
6.25.
Polyhedral representation of the three-dimensional framework in the crystal
structure of NiMn02(OH) shown along the X-axis as the packing of square pyramids [MeOS]
and octahedra [Me06], where Me
=
Mn or Ni. Oxygen atoms are located in the corners of the
polyhedra around the metal atoms. The latter are shown as large dark spheres.
'
RF
and profile residuals (Eqs. 6.18 to 6.22) can be easily lowered by increasing the number
of adjustable parameters in a completely unreasonable structural model. Thus, relevant
residuals should be employed to gauge the fit between the observed and experimental data
rather than as an exclusive measure of the rationality of the underlying crystal structure.
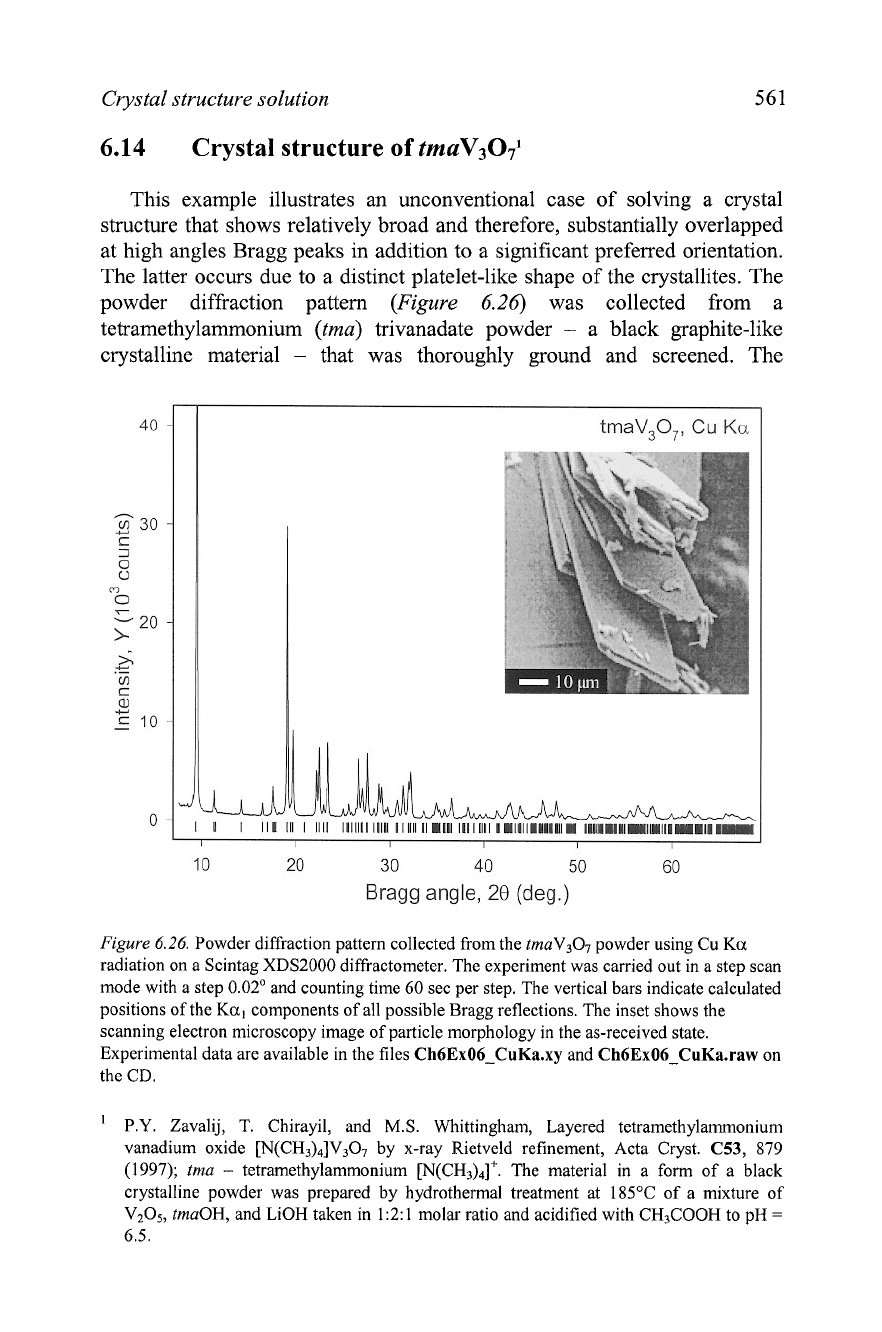
Crystal structure solution
561
6.14
Crystal structure
of
trn~V~0~~
This example illustrates an unconventional case of solving a crystal
structure that shows relatively broad and therefore, substantially overlapped
at high angles Bragg peaks in addition to a significant preferred orientation.
The latter occurs due to a distinct platelet-like shape of the crystallites. The
powder diffraction pattern
(Figure
6.26)
was collected from a
tetramethylammonium
(tma)
trivanadate powder
-
a black graphite-like
crystalline material
-
that was thoroughly ground and screened. The
I
10
20
30
40
50
60
Bragg angle,
29
(deg.)
Figure
6.26.
Powder diffraction pattern collected from the
tmaV307
powder using
Cu
Ka
radiation on a Scintag XDS2000 diffractometer. The experiment was carried out in a step scan
mode with a step 0.02' and counting time 60 sec per step. The vertical bars indicate calculated
positions of the
Ka,
components of all possible Bragg reflections. The inset shows the
scanning electron microscopy image of particle morphology in the as-received state.
Experimental data are available in the files
Ch6Ex06-CuKa.xy
and
Ch6Ex06-CuKa.raw
on
the
CD.
1
P.Y.
Zavalij,
T.
Chirayil, and M.S. Whittingham, Layered tetramethylammonium
vanadium oxide
[N(CH&]V307
by x-ray Rietveld refinement, Acta Cryst.
C53,
879
(1997); tma
-
tetramethylammonium
[N(cH&]+.
The material in a form of a black
crystalline powder was prepared by hydrothermal treatment at
185T
of a mixture of
V2O5, tmaOH,
and
LiOH
taken in
1
:2:
1
molar ratio and acidified with
CH3COOH
to
pH
=
6.5.